
Abstract
In liver tissue engineering, cell culture in spheroids is now well recognized to promote the maintenance of hepatic functions. However, the process leading to spheroids formation is time consuming, costly, and not easy to scale-up for further use in human bioartificial liver (BAL) applications. In this study, we encapsulated HepaRG cells (precursors of hepatocyte-like cells) in 1.5% alginate beads without preforming spheroids. Starting from a given hepatic biomass, we analyzed cell differentiation and metabolic performance for further use in a fluidized-bed BAL. We observed that cells self-rearranged as aggregates within the beads and adequately differentiated over time, in the absence of any differentiating factors classically used. On day 14 postencapsulation, cells displayed a wide range of hepatic features necessary for the treatment of a patient in acute liver failure. These activities include albumin synthesis, ammonia and lactate detoxification, and the efficacy of the enzymes involved in the xenobiotic metabolism (such as CYP1A1/2).
Impact statement
It has been recognized that culturing cells in spheroids (SPHs) is advantageous as they better reproduce the three-dimensional physiological microenvironment. This approach can be exploited in bioartificial liver applications, where obtaining a functional hepatic biomass is the major challenge. Our study describes an original method for culturing hepatic cells in alginate beads that makes possible the autonomous formation of SPHs after 3 days of culture. In turn, the cells differentiate adequately and display a wide range of hepatic features. They are also capable of treating a pathological plasma model. Finally, this setup can easily be scaled-up to treat acute liver failure.
Introduction
Fulminant liver failure is a life-threatening critical illness with an extremely high mortality rate, even if intensive care is provided. 1 To date, liver transplantation is the only treatment available for patients with end-stage liver disease such as acute liver failure (ALF). 2 The growing gap between the number of patients on waiting lists and the number of donor organs available has highlighted the need for alternative therapies, such as bridges to transplantation or liver regeneration. Extracorporeal liver support systems may be a valid solution for this issue, and today two types of devices are considered: artificial (ALs) and bioartificial liver support systems (BALs). ALs are designed to remove water-soluble toxins from the patients' plasma to improve their clinical condition, 3 while BALs are intended to fulfill the maximum number of synthetic and detoxification liver functions. 4 The key component of a BAL is the bioreactor, the cell-housing component. The role it plays is either to allow patients with ALF to survive until a liver donor becomes available or to stimulate autonomous liver regeneration.
Access to a bioactive mass (or biomass) is the main challenge in the context of human BALs. Although primary human hepatocytes are still considered the gold standard, their limited availability, the phenotypic instability, 5 as well as logistical issues, hamper their use in BALs. In the past, most of the BALs in clinical or preclinical studies relied on primary porcine hepatocytes, due to their easy availability and high functional activities.6–8 However, transfer of zoonotic diseases, 9 interspecies protein–protein incompatibility, and possible immune responses generated during treatment remain challenges for the use of xenogeneic hepatocytes. 10 Recently, the focus has been put on using induced pluripotent stem cells and embryonic stem cells as a source of innovative cells for BAL, especially for their considerable proliferative capacity and their potential to show metabolic functions similar to those of human hepatocytes. Despite great efforts in this direction, today these cell types are not yet capable of exhibiting the full functions of mature hepatocytes in terms of metabolic performance, and further efforts must still be made to make their clinical use possible. 11
Currently, human cell lines thus have great potential for BAL applications. The main advantages of hepatocyte cell lines are their almost unlimited proliferative capacity and the relatively cheap culture process. They demonstrate some metabolic functionalities comparable to those of human hepatocytes and are also safe and nontumorigenic. 12 HepG2 and its subclone present limited metabolic functions 5 for BAL applications, although encouraging results were recently presented by Selden's group on a pig model of ALF. HepaRG is a hepatic progenitor cell line capable of differentiating into hepatocyte-like cells after 14 days of specific culture. 13 Investigators have already demonstrated that the HepaRG cell line was suitable for BAL applications because of their capacity for proliferation and their enhanced hepatic metabolism in three-dimensional (3D) configurations. 14
In the context of BAL, hepatic cell microencapsulation in alginate porous beads has been recognized as an interesting alternative to classical cell immobilization in hollow fiber membranes. This material is quite inert, not toxic regarding cells, and ensures adequate biocompatibility. In addition, its relatively low cost and gelation capacity through divalent cations such as Ca2+ make the process suitable for hepatocyte encapsulation. 15 In vitro liver models combined with tissue engineering approaches provide a 3D microenvironment, which can be expected to mimic in vivo conditions. In the encapsulation process, cells are entrapped within spherical alginate microbeads that protect them from mechanical stress, while ensuring exchanges of nutrients or waste molecules within the surrounding medium. The immunoisolation provided by alginate encapsulation is undoubtedly the main advantage of this technology. 16
For BAL applications, the challenge is to induce/maintain the hepatic functions over time, starting from an initial biomass, taking into account parameters such as costs, ease of manipulation for the operator, and the feasibility of scaling-up. Recently, some authors successfully enhanced the biosynthetic and xenobiotic performances of HepaRG functions, cultivating cells as spheroids (SPHs) before encapsulation, without the use of dimethyl sulfoxide (DMSO). 17 Nevertheless, the technologies available nowadays for the production of large quantities of SPHs (such as rotating or shaking reactors) may lead to an unacceptable loss of cells, which considerably reduces the initial biomass. Most of these studies focus on toxicology approaches, in which it is important to evaluate the metabolic activity “per cell.” In the context of BAL, the challenge is not to determine the best activity “per cell” but rather “per bioreactor,” taking into account logistic hurdles or costs due to additional manipulation.
Therefore, in this study we suggest analyzing the cell differentiation and metabolic performances of an initial biomass of encapsulated HepaRG in alginate 1.5%, for further use in a fluidized-bed BAL.
Materials and Methods
Two-dimensional cell culture
HepaRG cells from Biopredic (Rennes, France) were expanded into two-dimensional (2D) monolayers following the indications given by the supplier. The cells were passaged every 2 weeks until passage 18, with William's E proliferation culture medium (WE, with sodium bicarbonate, without L-glutamine and phenol red, Sigma-Aldrich, with added Biopredic 710 proliferation media) replenished thrice a week. The cultures were maintained in a humidified environment at 37°C, 5% CO2. After that, the cells were detached with trypsin-ethylenediaminetetraacetic acid (EDTA) 0.25% (Thermo Fisher Scientific) from the culture flasks and used for the cell encapsulation process (explained in the following paragraph).
Alginate microencapsulation
The cells were encapsulated in alginate (Manucol LKX from FMC BioPolymer) 1.5% (w/v), sterilized with successive filtrations (0.8, 0.45, and 0.22 μm pore size membrane filters). The encapsulation was performed using a homemade system based on a coaxial air flow extrusion method. 18 Briefly, the alginate solution (1 mL) containing cells (5 million) was rapidly extruded through a 24 G nozzle, and the droplets fell into a gelation bath (NaCl 154 mM, HEPES 10 mM, and CaCl2 115 mM, pH 7.4). This low quantity of solution ensures even distribution of the cells throughout the beads and the absence of empty beads. The droplets produced were allowed to settle for 15 min in the gelation bath to ensure gel formation. After that, the microbeads were washed thrice in WE medium and then resuspended in proliferation culture media. Finally, the encapsulated cells were transferred to culture dishes and maintained for 14 days in continuous orbital shaking (60 rpm) in a humidified environment at 37°C, 5% CO2. The proliferation culture medium was replaced every 2 days. One milliliter of empty microbeads (without cells) was also produced as controls for the following metabolic tests.
Measuring cell aggregate diameters
The diameter of the cellular aggregates was determined by measurements made with ImageJ software version 1.52 h. For each experiment, the diameter of 10 aggregates in different beads was measured orthogonally by means of optical microscopy. Mean and standard deviation were calculated.
Cell performance experimental setup
The experimental setup designed to assess cell metabolic performance is shown in Figure 1. On day 0, the cells were encapsulated in alginate 1.5%. Cell differentiation was studied using immunofluorescence staining (see Immunostaining of hepatic markers) on days 0, 7, 10, and 14 postencapsulation, and the metabolic test (albumin synthesis) was carried out between days 0–1, 6–7, 9–10, and 13–14 postencapsulation. On days 7 and 14 postencapsulation, cell performance was tested by means of metabolic and xenobiotic tests. The latter were conducted at intervals of up to 2 h of incubation and, at each time point (time 0 and time 2 h), aliquots of supernatant were collected for further analysis. At the end of each experiment, microbeads were recovered and assessed for cell viability. The quantity of marker analyzed was calculated and normalized by the number of hours of incubation and the number of cells seeded. Moreover, to avoid artifacts, all the metabolic activities tested were normalized by the quantity present at time zero and by carrying out controls with empty microbeads. Therefore, any influence of the empty beads on the cellular activities assessed was corrected during the analysis of the results.
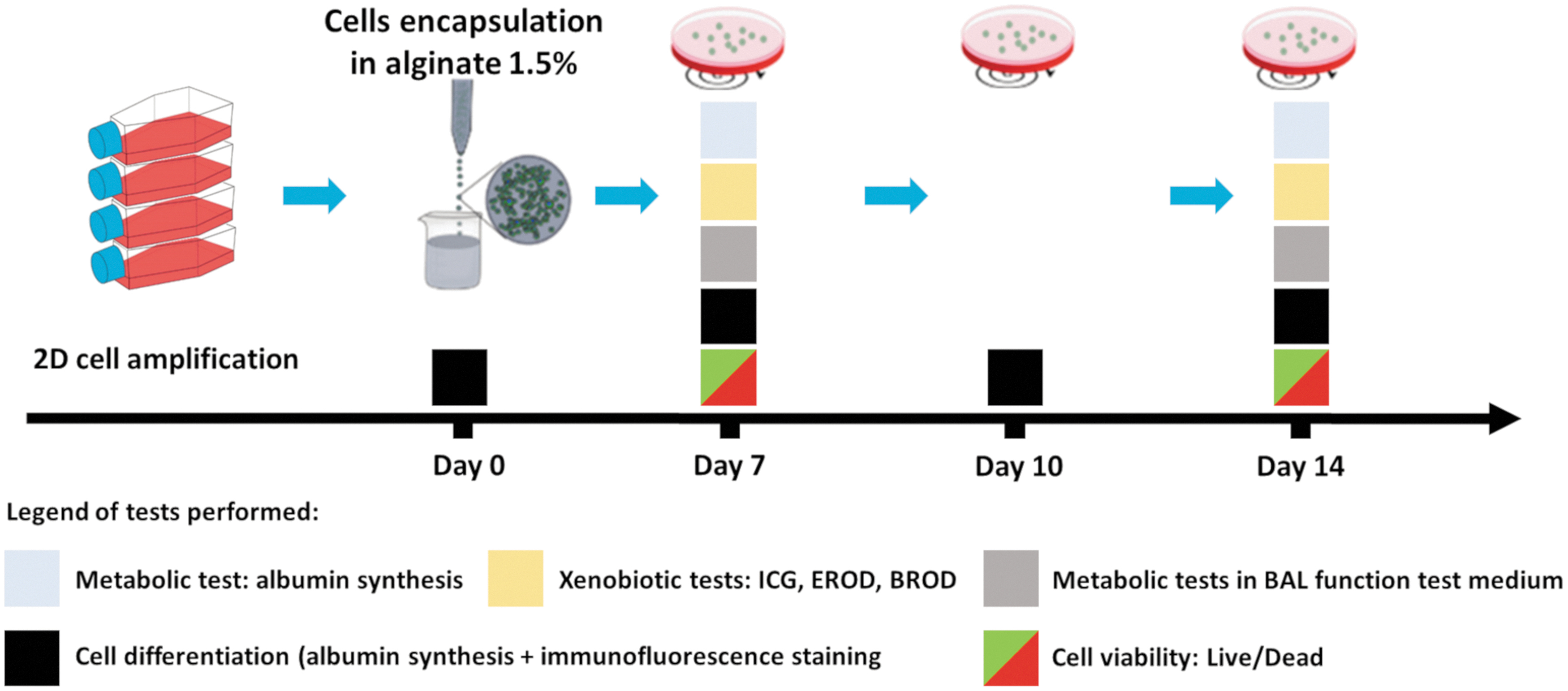
Experimental setup. After 2D amplification, cells are encapsulated in alginate beads and then cultivated for 14 days during which metabolic tests are performed. Color images are available online.
Metabolic and xenobiotic tests
Albumin synthesis
The test was performed in proliferation culture medium. Albumin synthesis was quantified using the ELISA test (Human Albumin ELISA Kit; Bethyl Laboratories, Inc.). Analyses were performed in conformity with the manufacturer's instructions.
Indocyanine green assay
As described by Gabriel et al., 19 indocyanine green (ICG; Sigma-Aldrich) was dissolved in DMSO at 25 mg/mL. Then, the solution was added to microbeads to obtain a total concentration of 1 mg/mL in proliferation medium. The microbeads were incubated for 30 min at 37°C and 5% CO2 to favor the uptake of ICG by the transporters OATP1B3 (solute carrier organic anion transporter family member 1B3) and NTCP (sodium/taurocholate cotransporting polypeptide). 20 ICG was then removed by washing with WE medium five times, and the microbeads were incubated with proliferation culture media for 1 h to study the release of ICG by the transporters MDR3 (multidrug resistance protein 3) and MRP2 (multidrug resistance-associated protein 2). 21 The quantity of ICG released was obtained by measuring its absorbance (at 820 nm) based on a linear standard curve generated from a known solution containing ICG 1 mg/mL.
Ethoxyresorufin-O-deethylase assay
The activity of the cytochrome P450 1A1/2 (CYP1A1/2) was studied with the ethoxyresorufin-O-deethylase (EROD) assay. 22 The microbeads were incubated in ethoxyresorufin (10 μM), prepared in WE, for 1 h at 37°C. The substrate included salicylamide (3 mM) + dicumarol (40 μM) to inhibit phase II enzymes. The substrate was converted into resorufin then measured in the supernatant by spectrometry (excitation wavelength of 535 nm and emission wavelength of 595 nm, Spectafluor Plus, TECAN). The standard curve was generated with a solution containing exogenous resorufin (10 μM).
In addition, the inducibility of the cytochrome was studied by adding β-Naphthoflavone 100 μM (Sigma Aldrich) and rifampicin 10 μM (Sigma Aldrich) to the media 72 h before the test.
7-Benzyloxyresorufin-O-dealkylase activity assay
The activity of the cytochrome P450 3A4 (CYP3A4) was studied with the 7-Benzyloxyresorufin-O-dealkylase activity (BROD) assay. 23 The microbeads were incubated in 7-benzyloxyresorufin (10 μM), prepared in WE, for 1 h at 37°C. The protocol was then the same as for the EROD assay.
Metabolic tests on bioartificial liver support system function test medium
Cultures on days 7 and 14 were incubated with proliferation culture medium containing 1.5 mM NH4Cl (Sigma Aldrich) +2 mM L-Lactate (Sigma Aldrich) described elsewhere. 24 After 2 h of incubation, the supernatants were collected and analyzed with Indiko™ (Thermo Fisher) to determine the concentration of ammonia, using the Lactate Assay Kit (Sigma-Aldrich) to measure the concentration of lactate, following the indications reported by the supplier, and with an ELISA Kit for albumin concentration.
Immunostaining of hepatic markers
On days 0, 7, 10, and 14 postencapsulation, aliquots of encapsulated HepaRG were degelified in a sodium citrate-EDTA solution (NaCl 154 mM, sodium citrate 50 mM, EDTA 55 mM, pH 7.4), fixed in 4% paraformaldehyde (PFA) in PBS for 4 h at 4°C, then cytocentrifuged (Shandon Cytospin®), and stored at −80°C. To detect intracellular epitopes, the cells were permeabilized with PFS solution (0.7% gelatine, 0.025% Saponin, 0.02% NaN3). Primary antibodies (Table 1) were incubated for 2 h at 37°C. Alexa Fluor 488-conjugated and Alexa Fluor 568-conjugated second antibodies were added and incubated for 1 h at room temperature. Cell nuclei were stained with DAPI (DAPI Fluoromount-G®), and phalloidin (Alexa Fluor™ 488 Phalloidin, Thermo Fisher) staining was used for F-actin visualization. Samples were visualized by means of confocal microscopy (Zeiss LSM 710).
List of Antibodies Used for Immunofluorescence Staining
Cell viability
To observe cell viability, at the end of each experiment, aliquots of encapsulated cells were collected and a Live/Dead assay was performed following the manufacturer's instructions (Invitrogen™ LIVE/DEAD™ Viability/Cytotoxicity Kit, for mammalian cells; Thermo Fisher). The nuclei were stained with Hoechst 33342 dye (3 μM). The encapsulated cells were visualized by means of confocal microscopy.
Data normalization
Metabolic activities are normalized by the quantity of metabolite produced or consumed/hours of incubation/millions of cells seeded. This way of normalizing data makes it possible to evidence overall cell activity starting from a given initial hepatic biomass.
Statistical analysis
All the results presented were obtained from at least three independent cultures (N ≥ 3). The statistical analysis was carried out using the software GraphPad InStat v.3.10. Unpaired data were subjected to a Mann–Whitney test, while paired data underwent a Wilcoxon matched-pair signed-rank test, with a 95% confidence level considered significant. p values are presented as follows: no stars: p > 0.05; *p ≤ 0.05.
Results
Morphology and viability of the encapsulated cells
Alginate beads containing cells were successfully produced. Qualitative observations using bright field microscopy showed homogeneous cell distribution among the beads (Fig. 2, “morphology”), without any visible empty beads. We counted ∼1100 beads per encapsulation batch (1 mL of alginate solution). As the initial biomass was 5 million cells/mL of alginate, cell density was ∼4500 per bead. The number of viable hepatocytes (in green) within the microbeads remained high over time, as very few dead cells (red stained nuclei) were observed, in comparison with the negative control where cells were killed by ethanol 70% exposure (Fig. 2). Therefore, the long-time culture did not affect cell viability.

Observation of bead morphology by bright field microscopy and cell viability (live/dead assay) by confocal microscopy. In green (Calcein AM): viable cells, in red (ethidium homodimer-1): dead cells, in blue (Hoechst 33342 dye): cell nuclei. Scale bar: 100 μm. Color images are available online.
Interestingly, after a few days of culture, the cells self-rearranged within the beads, forming compact aggregates. The evolution in their diameter over time is presented in Figure 3A, while the morphology of the aggregates is shown in Figure 3B, where an aggregate on day 14 was stained with phalloidin (F-actin staining).

Evaluation of the morphology of cells self-rearranged in aggregates.
Cell differentiation study
To study the differentiation of cells, on days 0, 7, 10, and 14 postencapsulation, the subcellular localization of proteins typically expressed by hepatoblasts and mature hepatocytes was assessed by immunofluorescence (Fig. 4). On day 0 postencapsulation, hepatoblasts were present as we observed their typical markers, such as AFP, HNF 6, and CK-19. During the differentiation process, the level of hepatoblast markers decreased, while the mature hepatocyte markers showed up (ALB, HNF 4α). CYP 3A4 was constantly expressed and functional on days 7 and 14. Those qualitative observations are summarized in Table 2. In parallel, synthesis of albumin remained stable overall throughout the 14 days of culture with a tendency to increase on day 10 (data not shown).
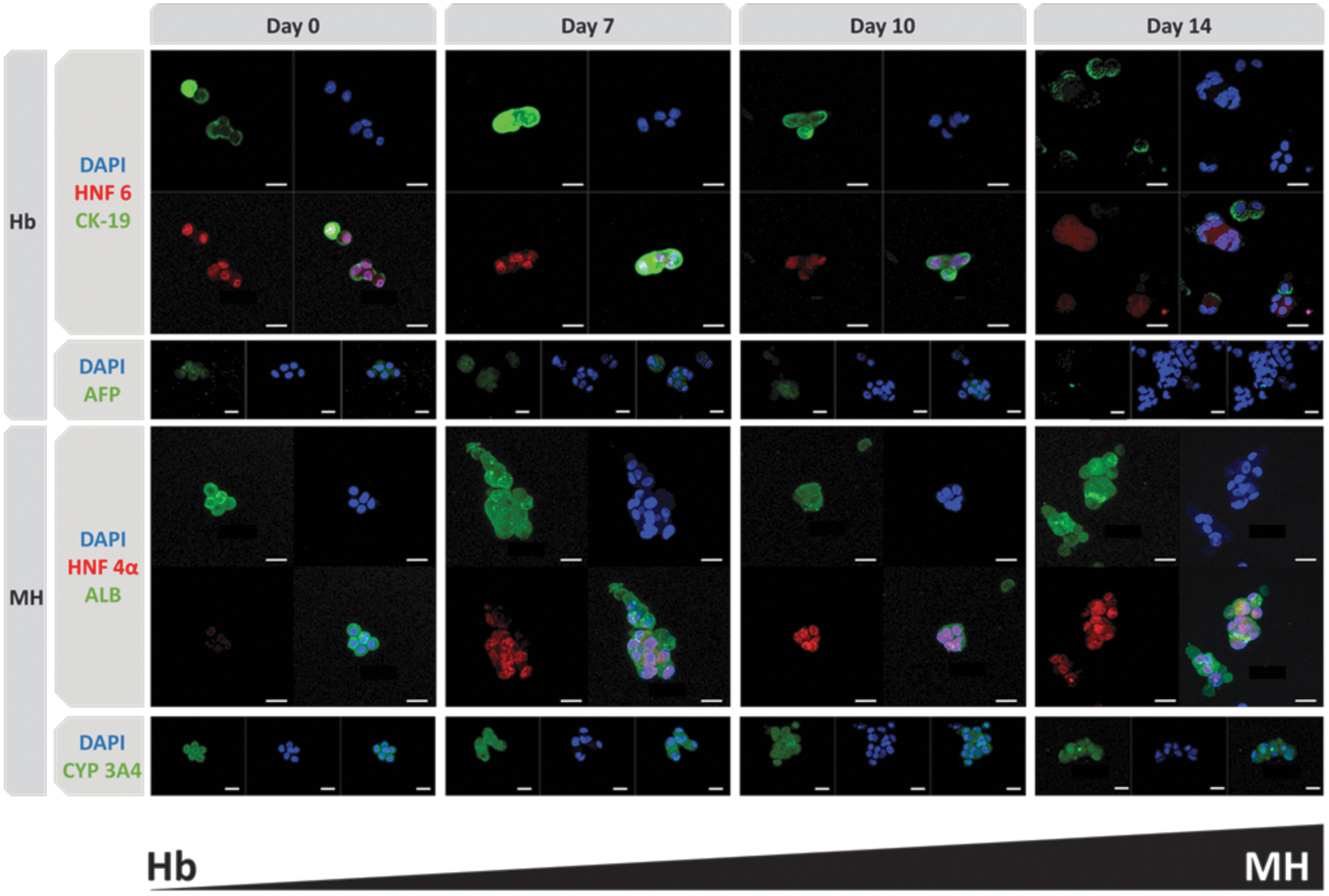
Immunofluorescence staining over time by confocal microscopy. Hepatoblast markers (Hb): HNF 6 in red, CK-19 in green, AFP in green, nuclei in blue. Mature hepatocyte markers (MH): HNF 4α in red, ALB in green, CYP 3A4 in green, nuclei in blue. Scale bar: 20 μm. Color images are available online.
Qualitative Evolution of Cell Differentiation Markers Over Time

Metabolic activity of cells on days 7 and 14
We compared the metabolic performance of encapsulated cells at 2 different days of differentiation, days 7 and 14. We analyzed the following metabolic activities: albumin synthesis (Fig. 5A), indocyanine green clearance and release performed by the enzymes involved in the xenobiotic metabolism pathway (MDR3 and MRP2 in Fig. 5B), CYP1A1/2 (Fig. 5C), and CYP3A4 (Fig. 5D). The results show that, with the exception of ICG, the cells had stronger metabolic activities on day 14, with statistical significance for albumin synthesis (1.35X higher). The basal activity of the cytochrome CYP1A1/2 was not detectable on day 7 but was measurable on day 14. Of note, CYP1A1/2 activity remained inducible through prototypical inducers on day 7 (Fig. 6A) and 14 (40X stronger, Fig. 6B). Overall, most of the metabolic activities analyzed tended to be stronger on day 14, most likely because the cells were better differentiated and rearranged into SPHs than on day 7. This statement is in accordance with the immunofluorescence staining shown in Figure 4.

Metabolic activities of encapsulated cells measured on day 7 and 14.

Measurement of CYP1A1/2 activity after induction by β-naphthoflavone 100 μM and rifampicin 10 μM.
Metabolic tests in BAL function test medium
Finally, we studied the ability of cells to detoxify typical ALF toxins, in view of a future application of these cultures in a BAL. The cells were incubated for 2 h in BAL function test medium containing ammonia and lactate. The ability of the cells to produce albumin and detoxify these toxins is shown in Figure 7. Again, the metabolic activities measured tended to be stronger on day 14. Importantly, the same samples tested on day 7 were tested again on day 14. After exposure to the BAL function test medium, the cell viability remained high and stable overall (Fig. 8).
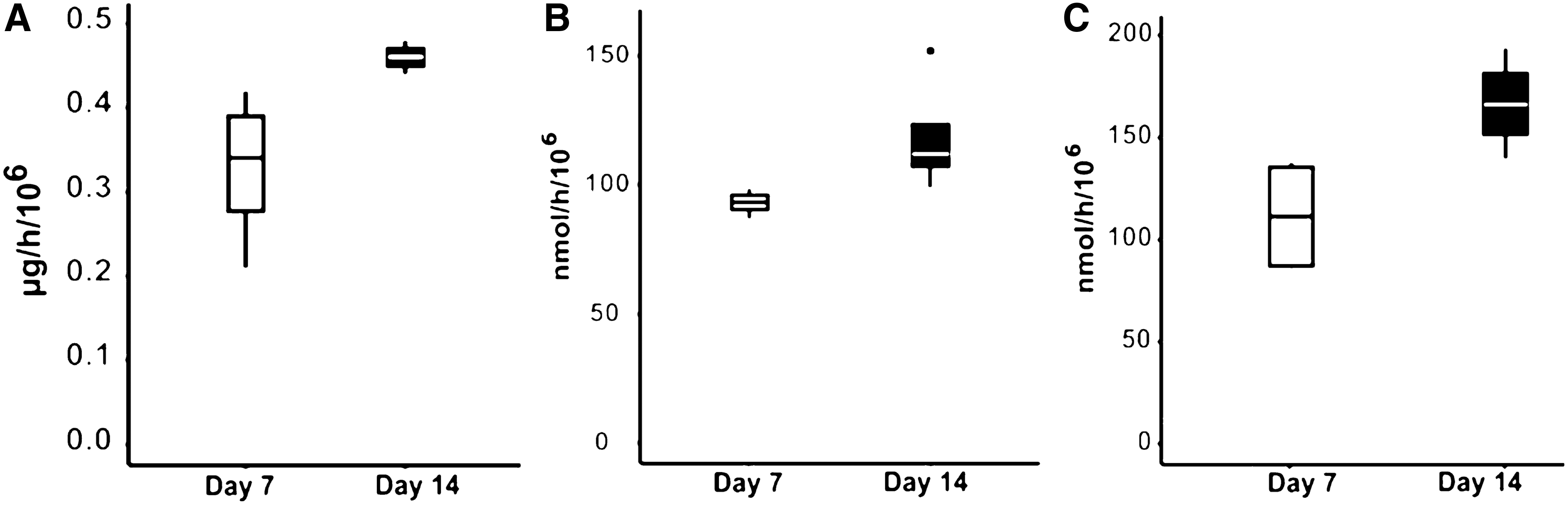
Metabolic activities of encapsulated cells measured in the “BAL function test medium” on days 7 and 14.
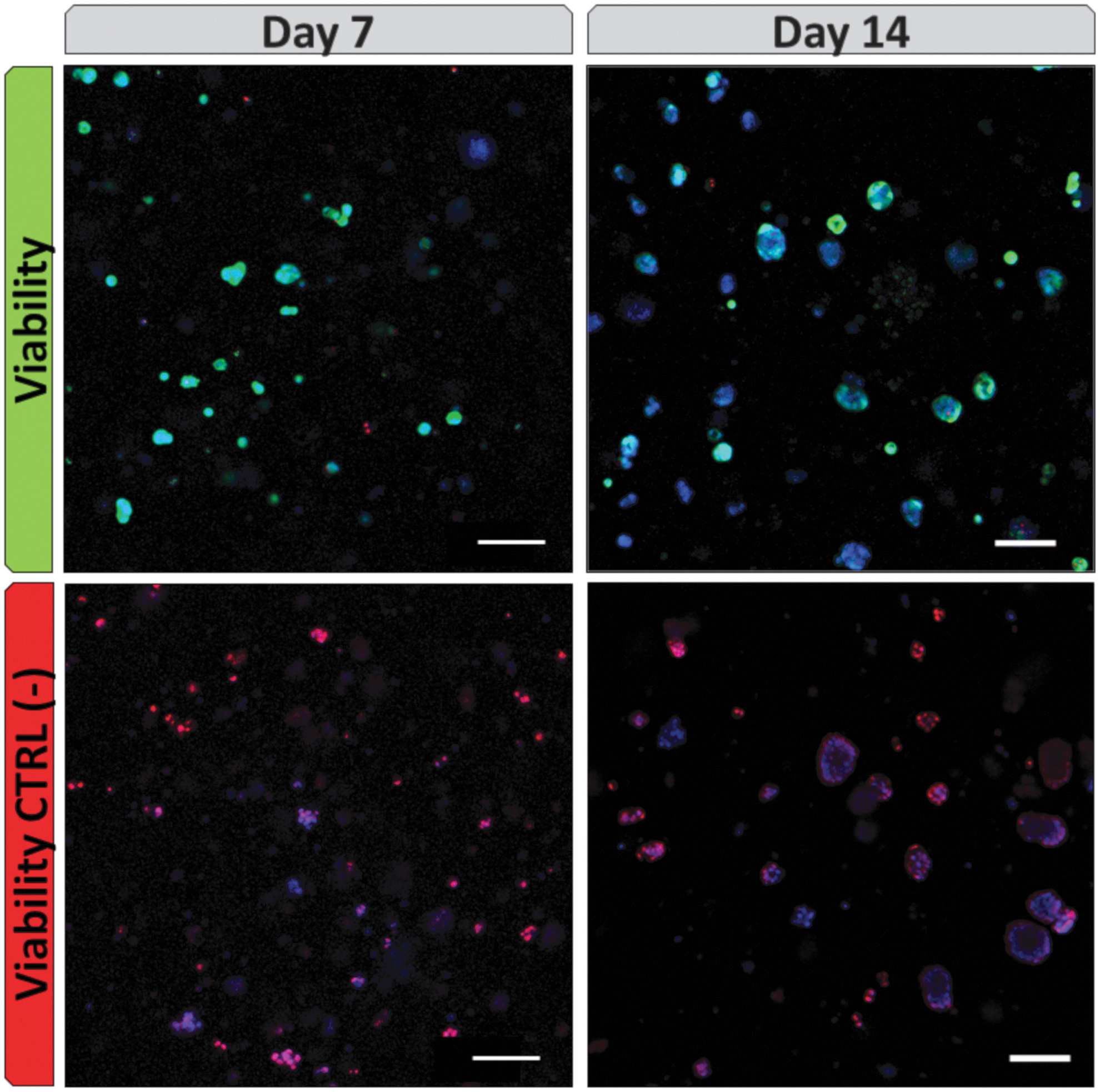
Cell viability (by live/dead assay) after exposition to “BAL function test medium” on days 7 and 14 by confocal microscopy. In green (Calcein AM): viable cells, in red (ethidium homodimer-1): dead cells, in blue (Hoechst 33342 dye): cell nuclei. Scale bar: 100 μm. Color images are available online.
It should be noted that the albumin synthesis rates in proliferation medium (0.64 ± 0.21 μg/h/106, mean and standard deviation, Fig. 5A) and in BAL function test medium (0.51 ± 0.08 μg/h/106, mean and standard deviation, Fig. 7A) were not significantly different. Therefore, the presence of typical ALF toxins (ammonia and lactate) in the medium did not seem to affect the protein synthesis activity.
Discussion
In this study, we analyzed the metabolic activities of a biomass of microencapsulated HepaRG for subsequent treatment of ALF. Such engineered liver tissues could be used in fluidized-bed BAL, a technology developed in our laboratory and in others. 25 With a similar bioreactor hosting HepG2 grown in alginate beads, Selden's group recently published the improvements to clinical parameters in a surgical porcine model of liver failure. 26 Zhou et al., also obtained prolonged survival time for pigs with D-galactosamine-induced ALF using a fluidized-bed BAL hosting porcine primary hepatocytes encapsulated in alginate-chitosan beads. 27
As stated in the introduction, further clinical applications require the production of large quantities of encapsulated cells to obtain a functional and cost-effective biomass at the human scale. The hepatic cell functions required are mainly protein synthesis (albumin and clotting factors), ammonia and lactate elimination (both implicated in hepatic encephalopathy and cerebral edema), and xenobiotic detoxification (enzyme machinery involved in cleaning toxins accumulating during ALF). 5 In the present study, we demonstrated that HepaRG microencapsulated in alginate beads was able to produce albumin, detoxify ammonia and lactate, and to express the enzymes involved in the xenobiotic machinery, with highest activity on day 14.
Our results show that cell viability was affected neither by the encapsulation process nor by the long cell culture time, nor by exposure to a toxic culture media, containing typical ALF toxins. Interestingly, within the alginate microbeads, the cells were able to rearrange, forming cell SPHs, a phenomenon already observed with HepG2/C3A and which are highly proliferative.28,29 In addition, mature hepatocyte markers were present, demonstrating that, when encapsulated, these progenitor cells were able to fully differentiate over time, in the absence of differentiation agents such as growth factors or DMSO, which are classically used in vitro. The 3D reorganization and alginate environment thus promote cell differentiation, in contrast with 2D culture.
Regarding cell functions, we performed a wide range of assessments. It is of note that encapsulated HepaRG was responsive to the ICG tests. Uptake of this dye is carried out by the transporters OATP1B3 and NTCP, which are expressed at the basolateral plasma membrane of hepatocytes 20 and excretion is regulated by the transporters MDR3 and MRP2, 21 expressed in the hepatocyte apical (canalicular) membrane. 30 In addition, F-actin accumulating at cellular junctional sites of the cell aggregates resembled interconnected bile canalicular structures. This observation was also noticed by Rebelo et al. Both facts indicate highly polarized cellular organization favoring cell maturation. 31 The dimensions of the cell aggregates increased over the days of culture but remained within a range (maximum 120 μm) where mass transfer was not affected and where the oxygen gradient could take place. 32 In addition, it has been observed that when HepaRG in a 2D configuration is cultured in orbital shaking, hepatic differentiation and metabolic functions are both positively affected due to higher mitochondrial biogenesis. 33 In our experimental setup, the encapsulated cells were also maintained in orbital shaking. These last two phenomena may induce a gain in function in the metabolic performances of the whole cell, which could explain why some of our activities were stronger in comparison with those of other authors.
Chamuleau's group seeded HepaRG in the Amsterdam Medical Center Bioartificial Liver (AMC-BAL), a membrane-based bioreactor in which cells are in direct contact with plasma. They reported ammonia (around 27 nmol/h/106) and lactate (around 10 nmol/h/106) detoxification with lower rates in comparison with our results (respectively, 119 ± 20 nmol/h/106 and 167 ± 24 nmol/h/106). These differences may be explained by the fact that in our system, the cells formed SPHs on the one hand and were protected from shear stress by the alginate structure, on the other. 34 Compared to HepG2/C3A, we can observe that albumin production was in the order of magnitude and that all phases of enzymatic biodegradation were present, although they were lower than those of primary human hepatocytes just after their collection. Considering Selden's promising results in preclinical studies with HepG2, the same could be expected with HepaRG. As a cell line obtained from hepatocarcinoma, the same protection should be envisaged for safe use in patients, such as the presence of filters downstream of the bioreactor. 26
Going back to the encapsulation process, Rebelo et al., also entrapped HepaRG in alginate beads, with high performances reported regarding biotransformation and interesting toxicological issues. However, they encapsulated preformed SPHs and not individual cells, as proposed in the present work. When encapsulating a biomass for long time cultures, it is important to consider different logistical parameters influencing the whole manipulation process for future scaling-up for clinical BAL applications. From our own experience (Supplementary Data), we observed a loss of cells when preparing aggregates/SPHs before encapsulation, especially using processes that can be upscaled, such as rotating or shaking reactors. This loss is limited with hanging drop or microfluidic techniques, 35 but the latter cannot treat billions of cells as needed for a human-size BAL. In addition, aggregate preformation requires at least 3 days of specific culture, before encapsulation, and additional handling that makes GMP production more complex and not cost-effective. As our preliminary results did not show any evidence of better performances of encapsulated preformed HepaRG SPHs (Supplementary Figures S1–S3), we concluded that their self-reorganization with alginate beads with appropriate density should be preferred in a clinical application strategy.
In conclusion, in this study we demonstrated that HepaRG cells were able to self-organize as SPHs when entrapped in 1.5% alginate beads. After 14 days of culture, these encapsulated cells presented a wide range of functions (protein synthesis, enzymatic activities, and biotransformation of toxins) that are within the range or above those presented in other studies on BALs. Waiting for fully mature hepatic cells issued from pluripotent stem cells, this HepaRG-based bioconstruct demonstrated its potential for further use for extracorporeal treatment of ALF. The next step will thus be to characterize biomass activity in fluidized-bed BALs, a setup known to favor mass exchange between the ALF patient's plasma and the immobilized cells.
Footnotes
Acknowledgments
The authors thank Baptiste Hirsinger, Marwa Hussein, and Augustin Lerebours for their technical assistance.
The HepaRG cell line, media, and supplements used for this investigation were supported by Biopredic International (funded by ANR 16-RHUS-0005 in the framework of the iLite programme), for which the authors express their appreciation. This cell line has been patented by Inserm, under number WO 03/004627. The authors acknowledge the English revision performed by Kirsty Snaith, MEDICIS TRADUCTION, Montpellier, France.
Disclosure Statement
There are no conflicting financial interests.
Funding Information
This work was supported by PIA-RHU iLite (ANR 16-RHUS-0005).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.