
Abstract
Dental caries have plagued humans for many years. At present, photocrosslinking resin is commonly used in clinics to repair narrow tooth defects, but the ultraviolet light used in this process has unavoidable cytotoxicity. In situ hydrogels with a similar structure to that of the natural extracellular matrix have gradually attracted attention in the field of hard tissue repair engineering. The injectable molding properties of hydrogel also give it the potential to fill irregularly shaped or fine tissue defects. Through a rapid and facile Michael addition reaction, we prepared maleic chitosan (CS-maleic anhydride [MA]) and thiolated alginate (sodium alginate [SA]-SH) to form a CS-MA/SA-SH hydrogel. To endue its mineralize ability, β-glycerophosphate calcium phosphate and calcium carbonate as the precursor of hydroxyapatite (HAp) were premixed in the hydrogel at certain ratios. This kind of hydrogel can quickly form into different shapes within 10 min. It is worth noting that external Ca2+ can react with the residual carboxyl groups of SA and provide the hydrogel with a self-healing ability. At the same time, we creatively propose a method that uses alkaline phosphatase to promote the mineralization of HAp in hydrogels, to achieve the purpose of regenerating hard tissue in situ. By examining the properties of hydrogels at different concentrations of calcium and phosphorus salts, we find that the CS-MA/SA-SH hydrogel with 50% (wt.%) inorganic matter presented the best self-healing properties, excellent mineralization of highly crystallized Hap, and has ideal cell compatibility. The potential application of the CS-MA/SA-SH hydrogel in repairing exposed dentin tubules in decayed teeth was explored through preliminary in vitro dental restoration experiments. Obviously, the penetration depth through dentin tubules was better than that of commercial dental sensitizers. In addition, the HAp morphology was affected by the local environment. We believe that this hydrogel can utilize tissues for dental regeneration and mineralization, and the healing ability provides the hydrogel flexibility for further application in hard tissue regeneration.
Impact statement
In this article, we report a simple, gentle, and rapid method for the synthesis of new, in situ-formable polysaccharide-based hydrogels that are capable of healing through the Michael addition reaction, which does not require any crosslinking agents. The rapid healing ability of the hydrogel can be obtained within 1 min by introducing calcium ions, giving the hydrogel the possibility of self-healing in the cap-enriched state. Second, we conferred in vitro mineralization of hydrogels and used a simple novel “enzyme-promoted” approach. Simulated mineralization using human body-enriched alkaline phosphatase as an inducer, allowing hydrogels rich in calcium glycerophosphate and calcium chloride to be highly similar to natural hydroxyphospholipids in a simulated body fluid environment at 37°C, stone-structured, and well-shaped thin-section mineralized products. Finally, we use the self-healing hydrogel with in vitro mineralization ability to carry out simple repair and mineralization regeneration of enamel and dentin, and this result is related to the tooth desensitizing agent commonly used in Chinese dental clinics. Comparison of commercial hydrogels further proves that this polysaccharide-based hydrogel is not only convenient for clinical operation in oral prosthesis but also has obvious advantages in the sealing of enamel by dental tubules and the enhancement of tooth hardness.
Color images are available online.
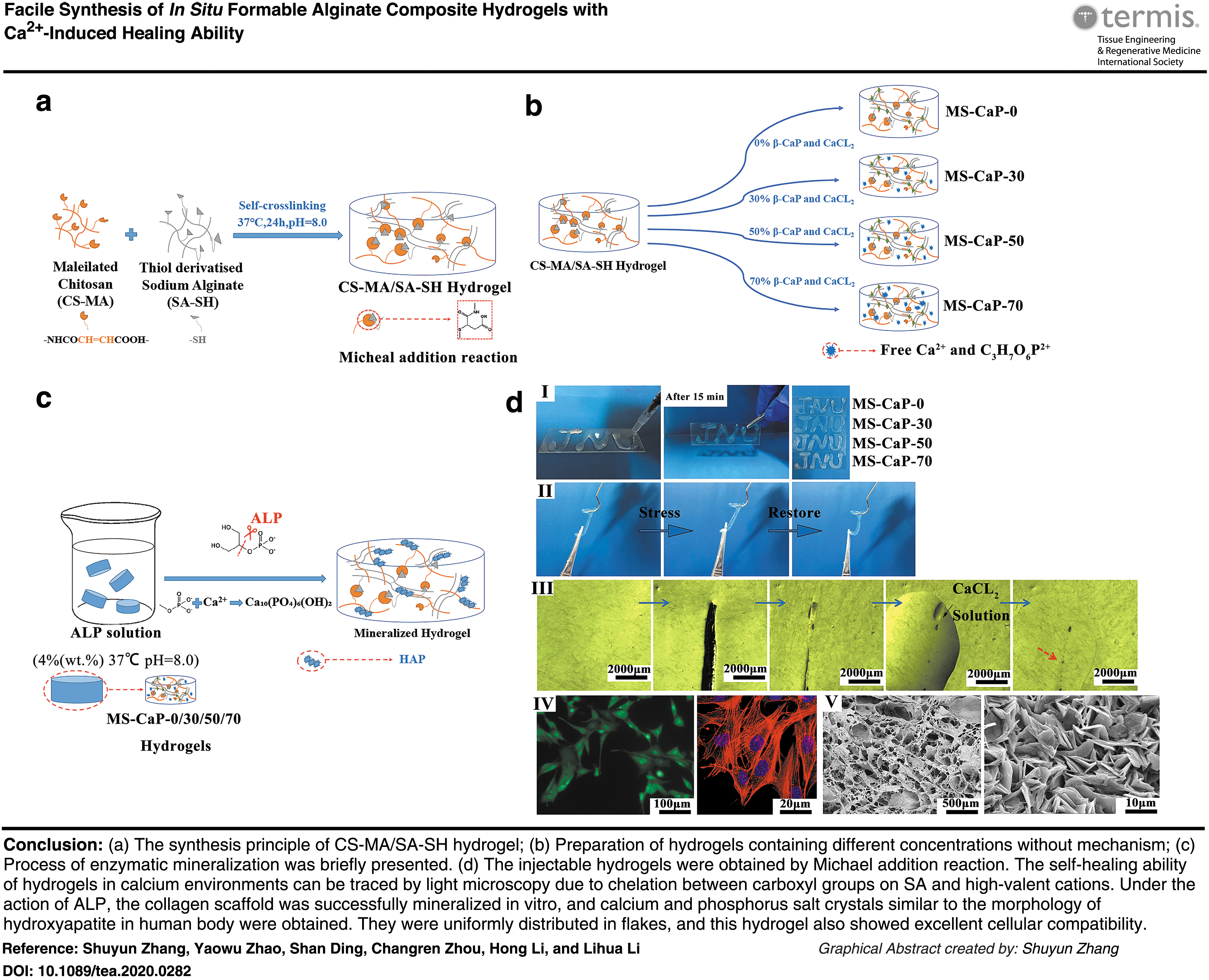
Introduction
Teeth are a combination of organic and inorganic tissue. The content of inorganic substances in teeth can be up to 96% or more,1,2 including amorphous calcium phosphate, octahydrate, dihydrogen phosphate, and hydroxyapatite (HAp).
With the development of medical science, autologous, allogeneic, and artificial materials have been used for the treatment of tooth defect. Common filling materials are mainly resins, metal powders, glass ions (acrylic acid and glass and fluoride) and ceramics. However, there are still many problems that cannot be ignored, such as donor deficiency, immune rejection, and inflammatory reactions. 3 Moreover, these materials do not have biological activity and can only play a role in filling and replacing cavities or tooth defects. Furthermore, making materials according to the shape of the defect undoubtedly increases the cost. Therefore, finding a highly permeable hard tissue repair material that can induce biomineralization and has shape plasticity is a potential goal for engineering tissue materials.
First, when choosing suitable materials, we found that the three-dimensional (3D) network structure of hydrogels endows them with excellent biocompatibility, cytoplasmic matrix-like properties, and little mechanical stimulation to human tissues. 4 These superior properties give it advantages in transporting nutrients, the release of growth factors, mobility, and the secondary molding properties. Due to hydrogels having these advantages, different ways to prepare hydrogels are applied in many aspects of the medical field.5,6 They use hydrogels for repair and for eliminating the need for a second surgery to remove implants.7–9
Hydrogels prepared by photocatalytic methods, pH response methods, and temperature response methods produce unpredictable cytotoxicity along with harsh reaction conditions that may induce other potential problems.4,10–12 Neverthless, the complexity of the synthesis, functionalization, and purification of hyperbranched macromonomers hinders the development prospects of these systems and increases the cost of production.13–15 In addition to excellent biocompatibility, mechanical strength, and cell biocapabilities, a good self-healing ability is also crucial for the application of biomedical materials.16–20 To date, although a few examples of tough hydrogels with self-healing properties have been reported, some of these healing processes use the hydrogen bonding, and covalent bonding of composite polymer materials21–25 and some of them require the application of an external stimulus such as heats26–28 and lights. 29
Second, it is essential to equip hard tissue repair materials with biomineralization ability. Alkaline phosphatase (ALP) is a type of nonspecific phosphomonoesterase that is widely distributed in human tissues. ALP can generate phosphate ions by hydrolyzing phosphocholine and phosphoethanolamine to increase the concentration of phospholipids.30,31 Phospholipids and Ca2+ have a strong affinity, so phospholipids and calcium ions can codeposit in the osteoid. 32 In current researches, ALP has been connected to fibrin, collagen scaffolds, and hydrogels to increase mineral deposits.33–35 However, commercial ALP is expensive and much will be lost when blending with materials. Therefore, we constructively propose that a certain proportion of calcium phosphate salt is added to the hydrogel, and the human body's own ALP is used to achieve the purpose of in-situ mineralization of the hydrogel.
Based on the survey and discussion above, in this study, chitosan (CS) and sodium alginate (SA), which are widely derived and highly biocompatible, were selected and modified, respectively.36–38
By the Michael addition reaction between modified products, CS-MA and SA-SH, gel sites are formed by addition reactions between the nucleophiles (e.g., thiol groups) and electrophiles (e.g., vinyl/acrylate groups),39–41
and a hydrogel material with a particular mechanical strength, excellent biocompatibility, and self-healing ability under the action of calcium ions is produced. It is worth mentioning that the CS-MA/SA-SH hydrogel can form a new crosslinking site under the ionic chelation of calcium ions and α-
This designed hydrogel can be used as an inorganic mineralization template. An ALP-induced calcium phosphate salt is placed between polysaccharides to mimic human hard tissue growth on the hydrogel network. The shearing effect of ALP separated a large number of free state phosphoric acid roots from glycerol phosphate.42,43 In addition, insoluble calcium phosphate salts, formed by reacting with calcium ions, precipitated and deposited in the stent. Then, the self-assembly method is used to create a solid biotissue-like inorganic layer. Thus, the potential of the hydrogel for biological hard tissue repair is explored.
Materials and Methods
Materials
CS (degree of deacetylation = 75–85%, viscosity = 200–800 cps, 1 wt.% in 1% acetic acid at 25°C [lit.]), maleic anhydride (MA), SA (viscosity ≥2000 cps, 2 wt.% in water at 25°C [lit.], the mannuronic acid/guluronic acid ratio of 1.56), and the calcium glycerophosphorate (Ca-GP) were purchased from Sigma-Aldrich. N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDAC), N-hydroxysuccinimide (NHS), beta-glycerophosphate disodium salt (β-GP), and calcium chloride anhydrous were of analytical grade and purchased from Qi Yun Biotechnology Co., Ltd. (China). The ALP was purchased from Huamaike Bio Co., Ltd. (China) and the commercial dental desensitizer Bgfresh from Hongyuan Medical Science and Technology Co., Ltd. (China).
Synthesis of maleic CS (CS-MA) and thiolated SA (SA-SH)
According to our previous work,36,44–46 CS was modified with MA to introduce vinyl carboxylic acid groups at the N-terminus of the CS to obtain CS-MA. In brief, 1.0 g CS was dissolved in 100 mL of 0.05 M acetic acid to form a homogeneous solution, and then 0.146 g of MA was added slowly afterward. The reaction mixtures were stirred in N2 for 24 h at room temperature, followed by 3 days of dialysis (molecular weight cutoff [MWCO] 14 kDa) with deionized water being replaced every 6 h. Finally, the resulting polymer solution was freeze-dried and stored at 4°C.
SA-SH was synthesized by the reaction of the carboxyl groups on SA with the amino groups of
CS-MA and SA-SH were characterized by nuclear magnetic resonance ( 1 H NMR), Fourier transform infrared spectroscopy (FT-IR), zeta potential, and ultraviolet (UV)–visible spectrophotometric measurements.
Preparation of polysaccharide hydrogels containing calcium phosphate
CS-MA and SA-SH were separately dissolved in deionized water. The concentration of SA-SH was 20 mg/mL, and the concentration of CS-MA was calculated according to the ratio of –C = C–/–SH = 1:1. Subsequently, Ca-GP and calcium chloride were dissolved in the CS-MA solution at 0%, 30%, 50%, and 70% (wt.%), respectively, with Ca/P molar ratio of 1.67. 1
Afterward, the above solutions were mixed evenly. The pH was adjusted to 7.0 with a 58% (wt.%) β-GP aqueous solution. Finally, the CS-MA/SA-SH gels were formed at 37°C and named MS-CaP-0, MS-CaP-30, MS-CaP-50, and MS-CaP-70 according to the amount of inorganic matter in them (refer “b” in Graphical Abstract for the sample preparation).
Rheological experiments were performed with a Kinexus Pro rheometer (Malvern, United Kingdom) using parallel plates (Ø 40 mm). The time-sweep of the precursor solution was carried out at 37°C, a frequency of 1 Hz, and a strain of 1% to determine the time of gel formation (denoted as the gelation time). Scanning electron microscopy (SEM; PHILIPS XL-30 ESEM, the Netherlands) was used to determine the morphology of the freeze-dried hydrogel. The swelling ratio (Q) of the hydrogel was determined as described in the literature 47 and defined as (Ws/Wd) × 100% (the weight of the swollen hydrogel/the weight of the dried hydrogel, n = 3).
In situ transformation of CaP in the hydrogels
ALP-mediated mineralization was used to simulate the mineralization of hydrogels in body fluids in vitro. To investigate whether HAp can be mineralized on the prepared hydrogel under enzymatic action in the human body, the freeze-dried gels were mineralized with a 0.4 mg/mL ALP 35 solution at 37°C and were then adjusted to pH = 8.0 with NaOH (1 M). After aging for 24 h, the samples were rinsed with deionized water to neutral and freeze-dried. Similarly, MS-CaP-50 was freeze-dried and mineralized in vitro for 12, 24, 48, 72, and 96 h, respectively. (refer “c” in Graphical Abstract for the sample preparation). The crystal composition of the samples was determined by X-ray diffraction (XRD; MSAL XD-2X Peking University Blige Technology Co., Ltd.) at a voltage of 36 kV, tube flow of 20 mA, CuKα radiation source, and scanning range (2θ) of 5°–60°, respectively. The morphology of all the samples was observed with SEM (working voltage of 20.0 kV).
Self-healing ability of the hydrogel
The secondary healing ability of the gels was evaluated as follows: hydrogels with different concentrations of calcium phosphate (Ø 15 mm) were cut into two equal slices, and then the rhodamine B-stained and unstained slices were alternately spliced to form a new cylindrical sample. CaCl2 solution (1 mL, 0.1 mol/L) was then added dropwise onto the surface of the recombinant sample (as shown in Fig. 5a). Tweezers were used to examine the healing effects after 10 min, and SEM was used to observe the morphology of the interface. (refer to Fig. 5a for the schematic diagram of sample preparation).
Oral implants are often subjected to external extrusion, so the compressive strength of the hydrogels before and after healing was tested with a universal material experiment machine (Shimadzu AG-1, Japan) to define the healing efficiency (HE). The samples (Ø 15 mm × 6 mm) were compressed at a constant strain rate of 2 mm/min at room temperature, and the compression strength of the hydrogels was recorded at 60% deformation (before healing) or the appearance of cracks (after healing). The HE was defined as the ratio of the compressive strength of the healed sample to that of its original counterpart.
Cell line and biocompatibility of materials
MC3T3-E1 cells (third generation) were cultured in Dulbecco's modified Eagle's medium containing 10% (v/v) fetal bovine serum and 100 U/mL penicillin–streptomycin solution in a humidified atmosphere (37°C, 5% CO2).48,49
After a culture period, 100 μL of the MC3T3-E1 cell suspension (3 × 104 cell/mL)was added to the MS-CaP-0, MS-CaP-30, MS-CaP-50, and MS-CaP-70 hydrogel samples in 24-well culture plates. Cells were also placed on a Petri dish without any material for use as control groups. The cells were cultured in an incubator (37°C, 5% CO2) for 1, 3, 5, and 7 days. Then, the culture medium was removed, and each well was gently cleaned three times with a phosphate-buffered saline (PBS, pH = 7.4) solution. Afterward, 1 mL of culture medium and 100 μL of CCK-8 reagent were added to each well. After culturing for another 3 h, the absorbance of the above medium was measured at 450 nm. The following equation was used to calculate the percentage of viability and the survival rate (n = 3):
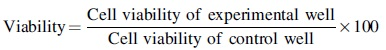
After culturing for 1, 3, 5, and 7 days, respectively, the culture medium was removed, and the cells were stained with acridine orange-ethidium bromide (AO-EB) staining solution (a mixture of 50 μL AO, 50 μL EB, and 5 mL of PBS) for 5 min. The cells were then thoroughly washed three times with PBS solution and observed by fluorescence microscopy (Nikon, Japan). After 3 and 5 days, the culture medium was removed, and the cells were stained with rhodamine-phalloidin (3% bovine serum albumin, 0.1% Triton X-100 PBS) at a ratio of 1:40 (200 μL/well). Then, the samples were stained with a 4′,6-diamidino-2-phenylindole staining solution prepared at a rate of 1:60 (300 μL, 10 min). After rinsing the sample three times with PBS, the cells were fixed using a 1:1 glycerol PBS solution. Finally, the sample was sealed for the subsequent observation of the cell growth morphology by laser confocal microscopy (LSCM).
Tooth defects repaired in vitro
Healthy teeth and dental caries were received from donors at the Overseas Chinese Hospital of Jinan University. Dentin cross section (D-CS), Dentin longitudinal section (D-LS), healthy enamel cross section (HE-CS), and molar enamel cross-section samples were sectioned and initially demineralized with an etchant (37% orthophosphoric acid). The treated tooth samples were sterilized in 5% (wt.%) sodium hypochlorite solution for 1 h and stored in artificial saliva (pH = 7.0, 14.4 mM NaCl; 16.1 mM KCl; 0.3 mM Cl2 · 6H2O; 2.9 mM K2HPO4; 6.16 mM CaCl2 · H2O; and 0.10 g/100 mL sodium carboxymethyl cellulose).
The MS-CaP-50 gel was uniformly cast on D-CS, D-LS, and HE-CS by syringe. The samples were placed in a constant temperature water bath at 37°C using an SBF aseptic simulated body fluid containing 4% ALP (wt.%). After aging for 48 h, the excess gel on the surface was washed away with an ultrasonic cleaner.
The D-LS samples were prepared and carefully ground to a thickness of ∼0.5 mm with a polisher. Afterward, the MS-CaP-50 hydrogel and commercial dental desensitizer were stained with 1 g/L rhodamine B and carefully cast into the D-LS cross section samples, while avoiding flowing into the longitudinal section. The samples were ultrasonically cleaned with phthalic acid and fixed. The penetration of the gel on the dental canal was observed and recorded by confocal microscopy. The surface morphology of the samples was observed using SEM (working voltage of 20.0 kV).
Results
Gelation of hydrogels
The chemical properties of MA-CS and SA-SH are provided in the Supplementary Data. The gelation of the MS-CaP hydrogels was confirmed by the vial tilting method, as shown in Figure 1a. The fluidic mixture transformed into a hydrogel within 15 min after homogeneously mixing CS-MA with SA-SH and β-GP, revealing the rapid formation of the hydrogel.
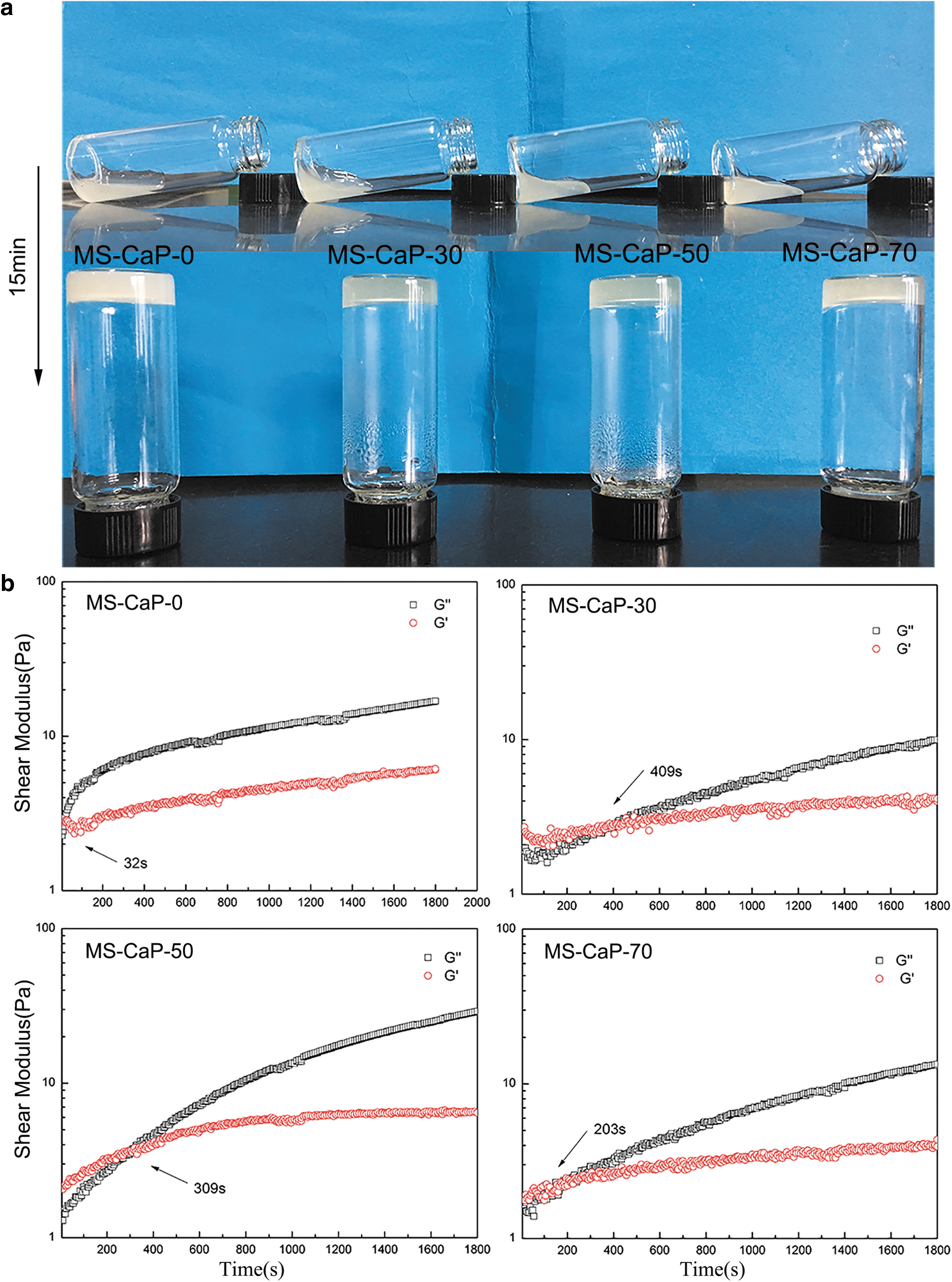
Gelation analysis of the MS-CaP gels.
The rheological behaviors in the time sweep are shown in Figure 1b. The storage modulus (G′) for MS-CaP-0 gradually increased and intersected the loss modulus (G′′) at 32 s. The gelation times for MS-CaP-30, MS-CaP-50, and MS-CaP-70 were 409, 309, and 203 s, respectively. Compared with the MS-CaP-0 sample group, the gelation time gradually increased and then decreased. With the addition of calcium ions, the gelation time increased. However, after increasing the calcium ion concentration, the gelation time is gradually decreased.
Morphology and swelling behavior of MS-CaP hydrogels
The microstructure of MS-CaP was observed by SEM, as shown in Figure 2a. All the samples displayed a continuous and porous structure due to freeze-drying. With the addition of calcium phosphate, the diameter of the pores decreased. Figure 2b shows that all the samples absorbed PBS rapidly within 30 min, then slowed down and gradually reached equilibrium. Sample MS-CaP-0 had the most significant swelling ratio, which was Qmax = 5.10 ± 0.15. With the increase in the proportion of CaP in the hydrogel, the pore diameter decreased, the water absorption capacity gradually decreased, and the Qmax of the MS-CaP-70 decreased to 2.30 ± 0.23.

Hydrogel surface morphology and swelling tests were carried out to confirm its water retention, which is an important guarantee that hydrogel can be used as biomaterials to provide nutrients and metabolic environment for cells.
Mineral transformation
The MS-CaP samples were checked with XRD, as shown in Figure 3a and b. All the samples except MS-CaP-0 showed typical HAp diffraction patterns after aging for 72 h, compared with the commercial HAp powders. The three peaks at ∼32° corresponded to the (211), (112), and (300) planes, the peaks near 46° corresponded to the (222) plane, and the peaks near 26° corresponded to the (002) plane; In addition, the increase in crystallinity was along the (211), (222), and (002) plane. Based on the results in Figure 3a, MS-CaP-50, the lowest concentration at which a characteristic peak appeared, was chosen to investigate the effect of time on calcium and phosphorus crystallinity. MS-CaP-50 showed the transition of Ca-GP and calcium chloride directly to HAp under the mediation of ALP in the solution. Figure 3b shows that the patterns of HAp in MS-CaP-50 became more evident with an increasing aging time.

X-ray diffraction patterns of MS-CaP hydrogels after mineralization.
According to Figure 3, after 72 h of aging, a more complete and obvious crystallization peak appears. We believe that if the crystallization process occurs, more mature HAp crystals can be observed. Therefore, we selected the hydrogel after aging for 72 h to investigate the effect of the calcium phosphate concentration on the crystalline morphology. According to previous experiments, we chose the lowest calcium phosphate concentration sample MS-CaP-50 with obvious characteristic peaks of HAp to investigate the crystallization time on the crystal morphology. The morphology of the MS-CaP hydrogels was observed with SEM, as shown in Figure 4 shows. Ca-GP and CaCl2 were embedded in the MS-CaP hydrogel as soluble salts and distributed uniformly inside. After aging with ALP, the calcium ions and phosphate ions were decomposed from Ca-GP, reacted, and later transformed to calcium phosphate salt in the MS-CaP hydrogels as time progressed. Therefore, the morphology of the hydrogels changed as a consequence. The MS-CaP-0 samples did not show deposits (Fig. 4a), and with the introduction of Ca2+ and PO43−, agglomerated inorganics appeared on the smooth network surface of the hydrogel. Notably, these agglomerated inorganics were not apparent at low concentrations (Fig. 4b, c), but rather when the calcium phosphate concentration increased. The salt agglomeration of granular or flower-like clusters was evident on the surface (Fig. 4d). Observing these details under the same Ca-P concentration and temperature, we found that the shape of these aggregates became more regular. The edge became thinner as the aging time increased (Fig. 4e–g). The shape of the inorganic mineralization gradually became regular and flaky (Fig. 4h), with a certain arrangement direction. These mineralized sediments were further scanned using energy-dispersive spectrocopy (EDS). Figure 4i–l corresponds to the calcium and phosphorus ratios (0.956, 1.199, 1.276, and 1.463) of the sedimentary inorganic matter at different mineralization times. In the preparation of materials, calcium and phosphorus in hydrogels are ionic. The concentration of calcium and phosphorus ions in the hydrogel material is relatively high, while the mineralization solution does not contain calcium and phosphorus ions. We believe that the calcium and phosphorus salts that do not form crystals will be separated from the material with the flowing mineralization solution and then washed away. The calcium and phosphorus contents observed in the subsequent EDS characterization should belong to the crystallization of a certain amount of calcium phosphate and slowly approaches the calcium-phosphorus ratio of 1.67 in natural HAp.

SEM images of hydrogel samples after enzymatic mineralization.
Ca2+-induced healing ability
In all experimental groups, two slices were integrated into a new cylinder for observation, and the interfacial parts could not be easily separated with tweezers 10 min later (Fig. 5b). With a light microscope, the healing process of the gel flakes could be better observed, and the wound was almost invisible after healing. When external forces were applied to both sides of the healed gel, the gel deformed but did not separate from the incision. Therefore, we believe that the gel has completed the healing process. This result revealed that there were still free carboxyl groups on SA in the hydrogels, which could be exposed due to cracks and react further with external Ca2+. Therefore, it can be concluded that after the hydrogel was implanted into bone defects full of Ca2+ for further application, it self-healed upon material failure. The compressive strength of the MS-CaP samples is listed in Table 1. With the introduction of CaP from 30% to 50%, the strength increased from 770 kPa to ∼900 kPa. The strength of all samples decreased after healing. The HE of the MS-CaP-0 hydrogel was ∼76.3%, and it decreased to 56.2%, 49.77%, and 43.70% with increasing concentrations of CaP.
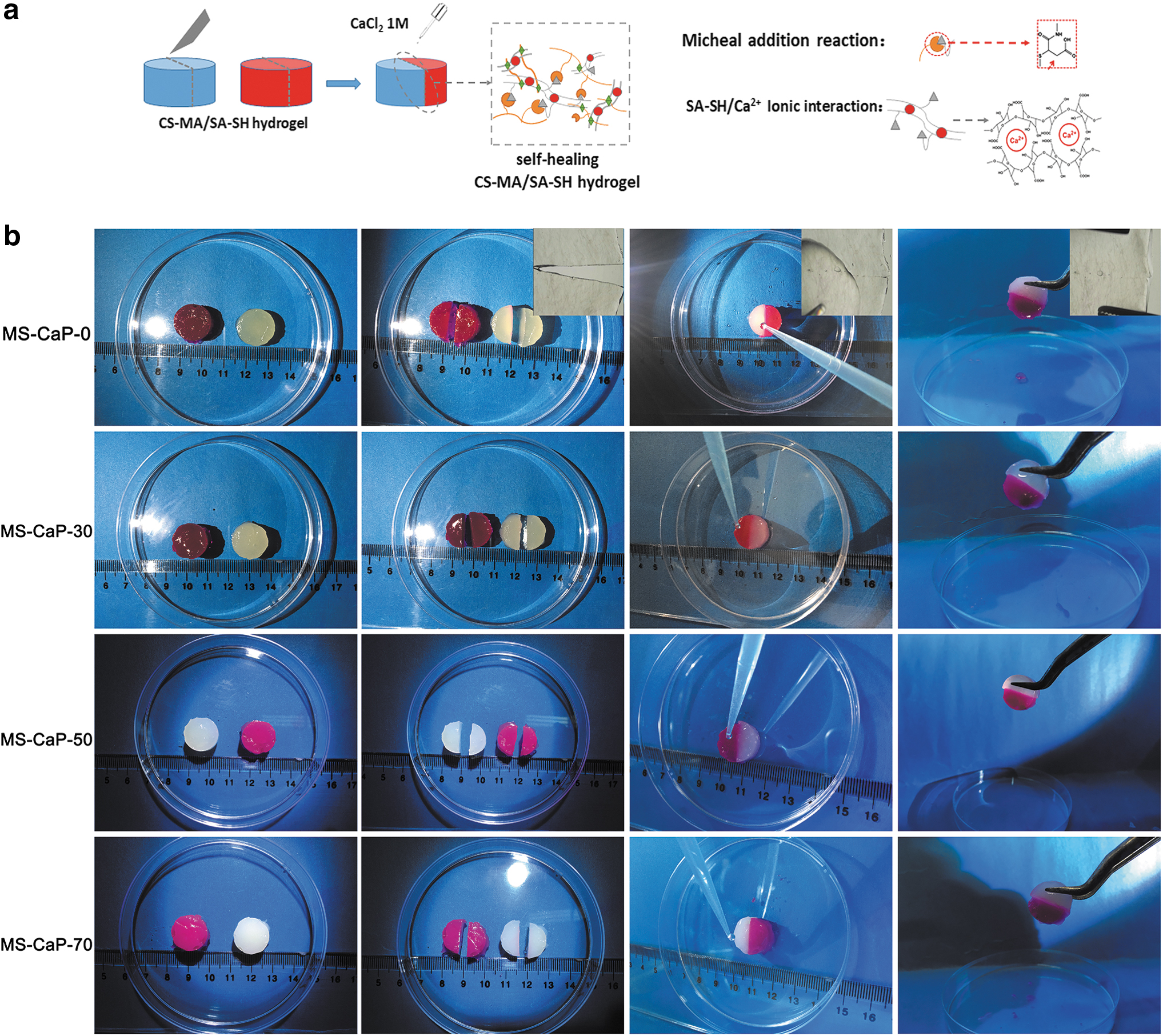
The self-healing property of hydrogel was verified by separation and recombination.
The Compressive Strength of the MS-CaP Hydrogels Before and After Healing
Data are expressed as means ± standard deviations (n = 3).
HE, healing efficiency.
Cytocompatibility of MS-CaP hydrogels
A fluorescence inversion microscope was used to observe cell activity (Fig. 6a). The number of cells increased with time for the experimental groups, although not as fast as the control. It is worth noting that the cells in samples MS-CaP-30 and MS-CaP-50 showed different degrees of aggregation in the first 3 days. Then, with the increasing inorganic content, the cells dispersed gradually. Simultaneously, the addition of CaP enhanced cell proliferation, and the cell number for MS-CaP-70 on day 7 was close to that of the control group. The change process of cells from long and narrow states to scattered and fusiform states was observed. LSCM was used to observe the morphology of the cells on the hydrogels with different calcium and phosphorus concentrations on the third and fifth days. The red parts showed a filamentous crisscross morphology, which was distributed in the cytosol and was the cytoskeleton. The blue part was the nucleus, and there was a tendency to stretch continuously. Consistent with the results of AO-EB staining, the cytoskeleton in sample MS-CaP-30 was constricted in the first 3 days. With increasing calcium and phosphorus salt concentrations, the cytoskeleton gradually spread out. By the fifth day, cells in all samples had shown the typical spindle shape of osteoblasts. Combined with the contents in section “Mineral transformation”, we preliminarily inferred that this result was due to the uneven distribution of inorganic matter in MS-CAP-30, and the low salinity and unevenly distributed sediment caused cell aggregation. With the increase in mineral content and the increase in mineralization, the coarse and uniform base was more conducive to cell migration to obtain surrounding nutrients.
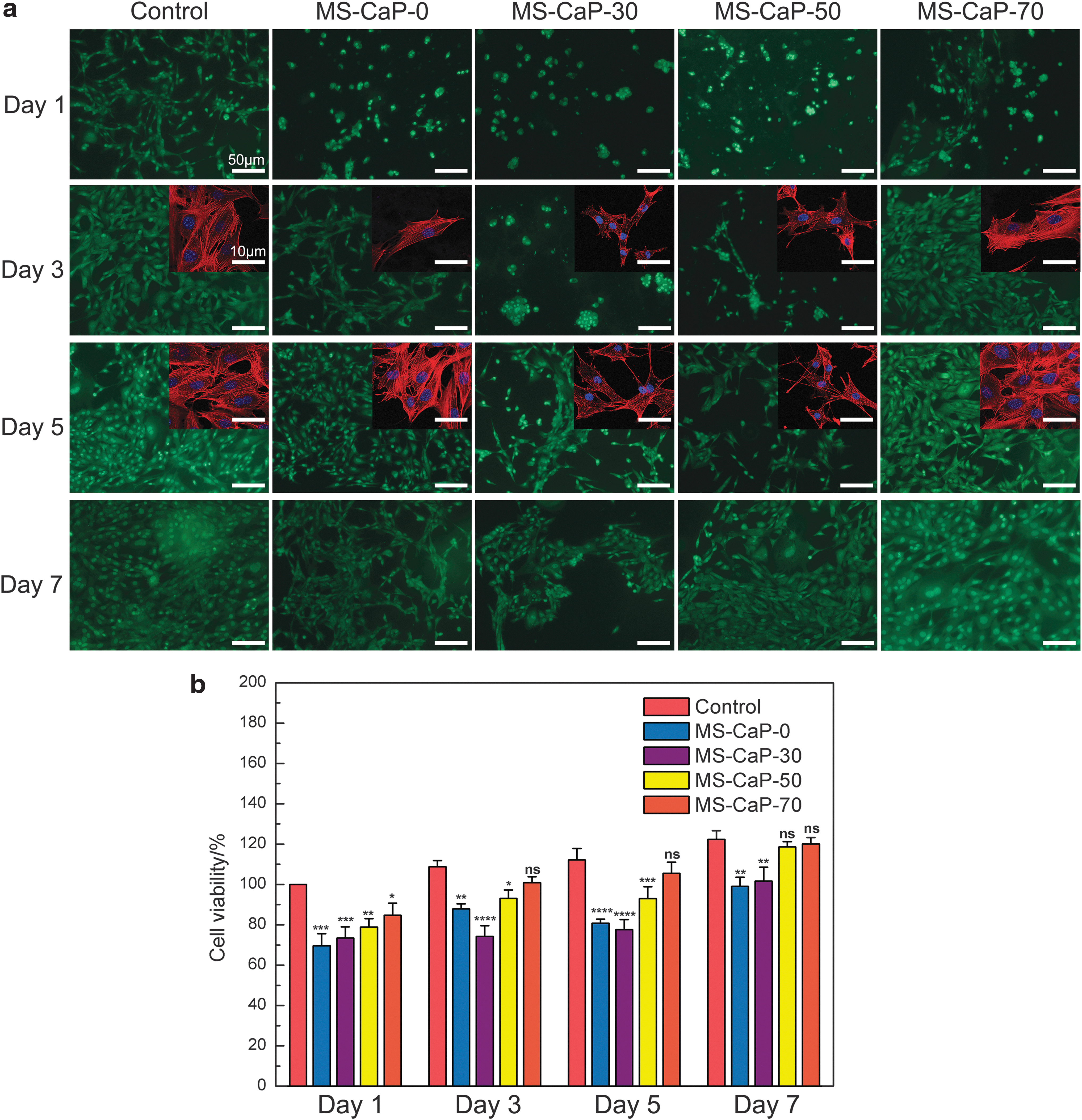
MC-3T3 cells were cultured on different hydrogels to test the cytocompatibility.
Cell viability was calculated according to GB/T 16886.5-2003 (ISO 10993-5: 1999) and the results are shown in Figure 6b. Clearly, except for the incubation time, the cell viability increased with increasing concentrations of CaP. In line with the AO-EB staining results, the MS-CaP-30 sample was not conducive to cell proliferation. In contrast, cell proliferation in samples MS-CaP-50 and MS-CaP-70 reached 97.1 ± 5.50% and 97.2 ± 2.49% on day 7, respectively, showing little difference from the control group. We preliminarily hypothesized that calcium phosphate ingredients could promote the proliferation of osteoblasts.
In vitro dental closure and repair test results
Since rhodamine B exhibits red fluorescence at an excitation wavelength of 546 nm, the red areas under confocal microscopy show the infiltration of the MS-CaP-50 gel and commercial dental desensitizer through dentin tubules (Fig. 7a). The commercial paste showed evident adhesion at the tooth surface; In addition, the penetration depth was ∼10 μm with an uneven distribution. In comparison, the penetration depth of MS-CaP-50 was 10–20 μm, with a higher density and uniformity. Figure 7b shows the mineralization of the enamel surface. The new mineral layer on the healthy tooth surface possessed a rod-like structure with a firmly arranged scale-like enamel structure. In dentin, the transverse and longitudinal sections of the tubules were filled at a high probability. The hydrogel underwent significant in vitro mineralization development on the dental canal sulcus, forming flaky and rod-shaped mineralized sediments.

The adult tooth was used as the defect sample to design the repair model after decayed tooth exposed tubule.
Discussion
Gelation of hydrogels
During the gel formation period, CS-MA and SA-SH formed an insoluble gel via Michael addition.41,50,51 The Michael addition reaction was confirmed by FT-IR, as depicted in the Supplementary Data. The characteristic peaks of the free unsaturated carboxylic acid group at 1691 cm−1, 1619 cm−1, and 897 cm−1 decreased and became a shoulder peak after reaction (indicated in the Supplementary Data). With the introduction of calcium, Ca2+ reacted rapidly with the carboxyl group of SA.52,53 Therefore, the Michael addition reaction between CS-MA and SA-SH was decreased, and the gelation time was delayed. However, with the continuous increase in the calcium salt concentration, the chelating effect between Ca2+ and SA-SH increased. Therefore, it would accelerate the gel formation of MS-CaP gels.
Morphology and swelling behavior of MS-CaP cryogels
A certain water retention is a common feature of hydrogel materials and the basis of their biocompatibility. Their porous structure provides a physiological environment for the metabolism of cells on materials. 54 With regard to MS-CaP hydrogel, with the addition of the calcium phosphate, the pore diameter decreased, perhaps due to two main reasons. Calcium phosphate in hydrogels affected ice crystal formation, and it is also blocked thermal transmission during the freezing process. Hence, the formation of the large layers was limited compared to that of the MS-CaP-0 samples. Inaddition, the chelating effect between Ca2+ and SA increased the crosslinking degree during gel formation, resulting in the formation of a denser structure. MS-CaP-0 had the most significant swelling ratio, mainly since the crosslinking density was the lowest, and the gel network was the loosest in all the samples. After introducing an ion reaction in the gelation process, the sample swelling rate gradually decreased with an increasing calcium-ion concentration. This result was due to the decrease in the hydrogel porosity, and swelling ratio results were consistent with the SEM results.
Mineral transformation
Studies have shown that glycerol phosphate calcium phosphate (GPC/Ca-GP) is an organophosphate with an anticaries effect. 55 Currently, it is widely used in in vitro biofilm research models 56 to enhance HAp resistance to demineralization and to protect enamel from the further demineralization of caries.57,58 Ca-GP can provide Ca2+ and PO43+ under the action of heat and the enzymes ALP for biomineralization under weak alkaline conditions. CaCl2 was used to supplement Ca2+, simulating the physiological environment close to the natural HAp calcium–phosphorus ratio of 1.67. ALP participated in HAp formation by hydrolyzing organophosphates and pyrophosphates, which are inhibitors of HAp crystal growth, to release phosphate groups, and this process was used to mimic slow in vivo biomineralization. Once the MS-CaP hydrogels were implanted into the body, the ubiquitous ALP in the body fluids would initiate the transformation of Ca-GP and CaCl2 to HAp.
Ca2+-induced healing ability
This self-healing hydrogel system imparted two-stage molding characteristics to the hydrogel: First, it was chemically crosslinked by Michael addition to obtain an in situ shaped primary shape. This phenomenon will expand the application prospects of these hydrogel materials in narrow topographical irregularities. Second, Ca2+ chelated with two carboxyl groups on the G-terminus in SA, giving the hydrogel a self-healing ability in a calcium-rich environment. The mechanical analysis results showed that the self-HE of the hydrogel decreased with an increasing Ca2+ concentration. The possible reason is that free Ca2+ occupied the G-terminal chelation space required for healing. These results further confirmed that the concentration of inorganic phosphate added to the CS-MA/SA-SH hydrogel should be between 50% and 70%, thereby meeting the conditions required for mineralization, and maintaining the excellent characteristics of the hydrogel.
Biocompatibility of MS-CaP hydrogels
From the analysis of cell activity assay results, MS-CaP-0, MS-CaP-30, MS-CaP-50, and MS-CaP-70 hydrogels have good biocompatibility. However, the low concentration of inorganic hydrogel was quite different from the control group. With an increasing of calcium phosphate concentration, the cell proliferation rate in the hydrogel samples was almost indistinguishable from that of the control, which indicated that the introduction of inorganic substances to the hydrogel materials did not promote cytotoxicity. The applicability of this material in a calcium-rich environment was further confirmed.
In vitro dental closure and repair test results
The results showed that the flow dynamics of the hydrogel to the solidified state could help it load and evenly distribute calcium and phosphate salts deep into the dentinal tubules for a more thorough and comprehensive tubular closure to protect the exposed dentin layer.
According to the Gibbs free energy, one reason for the formation of the rod-like structure may be due to the scaly structure of healthy enamel, which induced minerals to grow along the gully. From this point of view, crystal formation would gradually stack along with the more marginal parts. Another reason is ALP regulation, which induced the crystallization of calcium phosphate on the enamel layer and was stacked in a very regular arrangement. Similar results were obtained in the other two groups. The successful closure of the tubules demonstrated that the material could be used to repair the exposed dentin to fill the defect and to close the tubules to reduce pain. In addition, the damaged mineral layer was regenerated to prevent further damage to the pathogenic bacteria from eroding the dentin and pulp.
Conclusions
In this study, we prepared an in situ formable MS-CaP composite hydrogel through mild and rapid reactions. In this 3D network-structured hydrogel, β-CaP could transform into homogeneous HAp nanocrystals under ALP mediation in the human body. MS-CaP exhibited its optimum self-healing, biocompatibility, and biomineralization performance when the inorganic concentration was 50% (wt.%). In vitro dental closure and repair tests verified the suitable flexibility, penetrability, and gelation time of the gel precursors. Furthermore, MS-CaP hydrogels loaded with inorganic substances could penetrate the narrow tubules, and utilize the enzymes in the human body for in-situ remineralization. As an answer to the question mentioned at the beginning of this article, we found a hydrogel that could be easily prepared and could solidify quickly without a curing agent or UV light, making it suitable for clinical operation. This hydrogel can also be injected deep into an irregular tooth defect site and mineralized in situ from a research point of view. The healing ability of this hydrogel will enhance its application in Ca2+-rich hard tissue defects. Above all, the properties of the composite hydrogels identified in this study are beneficial for tooth repair applications, especially for hard tissue repair with a narrow and irregular topography.
Footnotes
Acknowledgments
The work is supported by National Key Research and Development Project (019YFD0901905), National Natural Science Foundation of China (31971284), Science and Technology Central Scientific Research Business Fee (11617458), Fundamental Research Funds for the Central Universities (21617424), and China Scholarship Council (File No. 202006780028).
Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by National Key Research and Development Project (019YFD0901905), National Natural Science Foundation of China (31971284), Science and Technology Central Scientific Research Business Fee (11617458), Fundamental Research Funds for the Central Universities (21617424), and China Scholarship Council (File No. 202006780028).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.