
Abstract
The fields of tissue engineering and regenerative medicine have the capacity to substantially impact clinical care through the introduction of new products that can address unmet clinical needs, or significantly improve on present therapies. These products will be developed through the demonstration of therapeutic effectiveness, adequate safety, and meeting regulatory requirements. The technology used in the product will dictate the product development and manufacturing costs; the regulatory pathway; and the time taken to complete clinical trials, gain regulatory approval, and become commercialized. A comparison of the required investment of time and funds, with the potential revenue generated, allows for a determination of the likely commercialization opportunity. Ultimately, the long-term success of a product will be dependent on its clinical effectiveness and commercial viability.
Introduction
Critical and necessary features of long-term product success include therapeutic effectiveness and profitability. 7 The introduction of these new technologies has been commercially challenging, with high product development and manufacturing costs, and slow market acceptance. However, recently, several of these products have moved into profitability. Both Dermagraft and Apligraf, both used for treatment of chronic wounds, are now profitable, although both had to pass through bankruptcy to get to that stage. 8 Perhaps most impressive from a commercial point of view is Infuse, 9 used to induce spinal fusion, and this serves as the shining example that new technologies can not only address clinical needs but also yield substantial commercial success.
The lessons learned during this translational phase of technology
8
are mostly obvious but are still frequently undervalued. They may include
• Modest investment until the major technical hurdles are overcome • Use of efficient manufacturing methods • Possessing robust business plans with realistic timelines, costs, addressable market size projections, and revenues.
When the products generate revenues that may be regarded as modest on a pharmaceutical scale (e.g., less than $100 million), the investment needed for bringing the product to the market becomes particularly important, because an appropriate return on the investment may be difficult to achieve. In any case, there should be a thorough assessment of the risks and benefits, and a balance of the challenges and opportunities should be reached. Issues to consider include
• Total cost of developing the product, gaining regulatory approval, and marketing to profitability • Time to profitability • Technical difficulty • Clinical uncertainty in outcome for the target patient populations • Regulatory challenges • Predicted market opportunities • International market opportunities and challenges
Therapeutic effectiveness is a general term used in this article to describe the clinical performance of any product, whether it is a device, biologic, or drug. Effectiveness of a device and efficacy of a biologic or drug are terms often used in regulatory discussions. New products may address previously unmet clinical needs, or more modestly only have similar clinical performance as products already in the market. In those circumstances, there are opportunities for products to be distinguished from competitive products by providing other advantages, for example, ease of use. Indeed, this type of competitive approach frequently occurs in orthopedics, ranging from bone fillers to joint replacements.
At the present time, the level of potential revenue and profit that may be achieved with most of these technologies and their applications will most likely be modest. Therefore, the quality of the strategic plan matched with effective execution will be a major factor that will determine the commercial success of the product, a prerequisite for long-term overall success of the product.
Product Development Pathway
The total product development pathway for almost any product involves a series of connected, overlapping but relatively distinct activities (Fig. 1). The project first requires the conceptualization of the product, followed by a series of research and feasibility studies, which may be in vitro or modest in vivo studies. These studies will test the concept, investigate mechanisms of action, may include optimization studies, and tend not to have specific timelines. An initial review of the business opportunities should indicate the potential for successful commercialization, and together with the results of the research studies, there may be the rationale for embarking on a defined product development pathway.
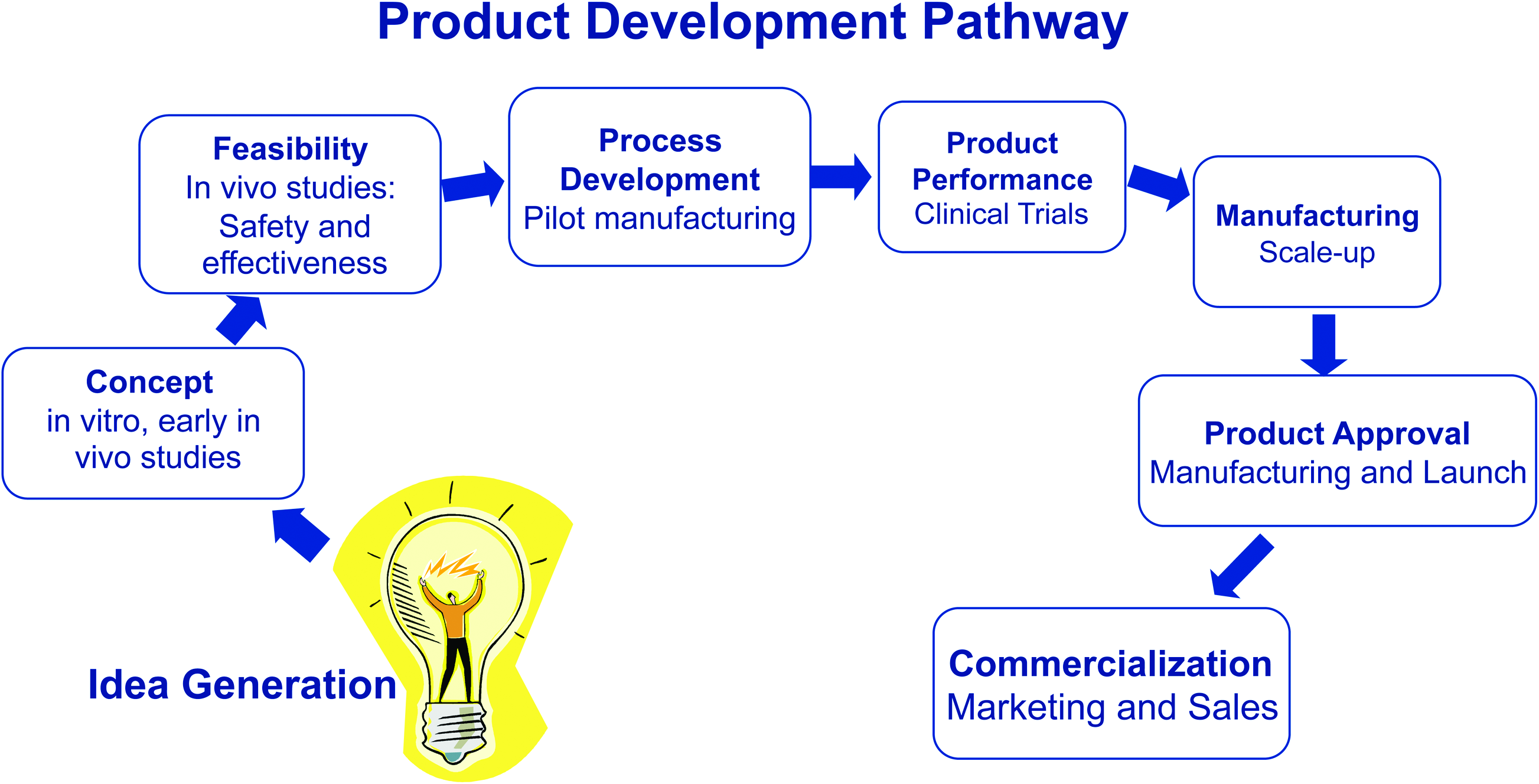
The diagram indicates the general stages that are encountered when developing a product, from the idea generation through to commercialization. Each stage may be considered distinct, but the activities may overlap in time. An umbrella activity ongoing during all of these is the regulatory process that is involved at all stages after feasibility studies are completed. Color images available online at www.liebertonline.com/teb
The next stage is to perform the key in vivo animal studies to test the therapeutic effectiveness and safety, 10 and these will be used as rationale for approaching the FDA or other regulatory authorities to gain permission to initiate clinical studies. They mark the beginning of a defined pathway of product development with stages involving defined activities, and will include pilot-scale manufacturing, clinical trials, scale-up to production-level manufacturing, regulatory approval, product launch, and commercialization. The project is reviewed multiple times to determine whether particular milestones have been reached to motivate moving to the next stage in the product development pathway. The complete process is managed in part by a quality system and standards that are required to be met by the FDA and any international regulatory agencies. If international sales is anticipated, then it is expeditious to consider using a quality system and a manufacturing system that meets the requirements of all agencies to be encountered.
The progression along that pathway is tied to a reduced technical risk of the complete project, but at the same time, there is a requirement for increased financial investment, because the activities (manufacturing, clinical trials) are more expensive. The decreased technical risk is, therefore, associated with an increased financial risk. The specific product development pathway activities are very dependent on the regulatory pathway that should be followed as required by the regulatory agency.
Regulatory Pathways
Within the United States, medical products are regulated by the FDA. There are three main pathways; 510k, device, and biologic/drug. The medical devices (including the 510k regulated products) are regulated within the Center for Devices and Radiological Health (CDRH), and the biologics and drugs are regulated with the Center for Biologics Evaluation and Research (CBER). The products in the fields of tissue engineering and regenerative medicine frequently involve multiple technologies that may be associated with more than one center within FDA. For example, the Infuse product has both device and biologic components. In these cases, the application to the FDA is first considered by the Office of Combination Products, where a primary assignment to a Center is made based on the “primary mode of action” of the product. The key steps and associated times and costs involved in each of these pathways are shown in Figure 2.
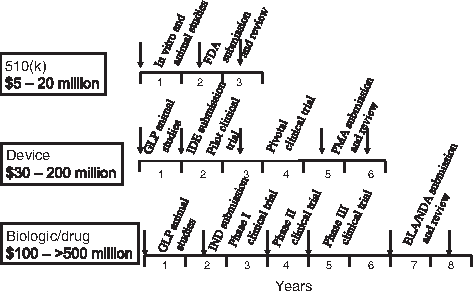
Approximate timeline and cost for the development of tissue engineered/regenerative medicine products in the orthopedic field, when regulated by the FDA as a 510k device, a device, or a biologic/drug. These timelines and cost estimates assume that all feasibility studies have been completed, and that the preclinical and clinical studies progress without major interruptions. IDE, investigational device exemption; IND, investigational new drug application; BLA, biologics license application; NDA, new drug application.
The 510k pathway involves the shortest time and the least development cost, but it relies on a restricted criteria dependent on the product being a device that can be shown to be substantially equivalent to another device which had been marketed before 1976. In reality, 510k products will be devices, and will be acellular; innovative products are unlikely to be regulated in this way, more likely the products will show only incremental improvements compared with previous products. The completion of the studies required to gain 510k regulatory clearance can usually be expected to take a minimum of 2–3 years, with additional time required at the beginning of the development pathway to develop the product concept to the stage where it is regarded as ready to be tested in major animal studies, if these are necessary. Resources will also be necessary to manufacture the product, and provide the infrastructure necessary to support this program. Examples of products that are regulated as 510k products range from bone cements to hip prostheses.
New medical devices are likely to be regulated through an investigational device exemption and premarket approval (PMA) regulatory pathway. The project will require major animal studies, and at least two clinical trials. Consequently, the cost of product development will be substantially higher than for a 510k pathway, and the time to market will be substantially longer, estimated to be a minimum of 5–6 years for most orthopedic products, from the time of initiating the key animal studies. Examples of devices that have gone through this pathway include disc prostheses (e.g., the Bryan Cervical Disc by Medronic). Augment by Biomimetics was designated as a combination product and was reviewed as a PMA.
A product where the primary mode of action is biologic (e.g., the product involves a growth factor, a drug, or cells) will use a biologics license applications or investigational new drug regulatory pathway. The pathway will involve animal studies and probably a series of three clinical trials. Consequently, this is the most costly and lengthy of the regulatory pathways, probably taking 8 years or more. Examples will include most cell-based products, and, therefore, is of particular interest to the tissue engineering and regenerative medicine fields.
The time and cost estimates for each of these pathways shown in Figure 2 are only approximations, and will be dependent on many different factors, including the specific product being investigated, the specific clinical treatment under investigation. There are examples of shorter times and less cost to get to the market; however, a more likely deviation from these estimates is longer time and/or increased cost. If the product is regarded as a combination product, then there may be additional tests and costs and potential for an extended timeline.
Return on Investment
A key feature in understanding the product development opportunities is to assess the market potential and likely revenue. In this business development activity, the market size should be estimated, the market penetration should be estimated, and the time to achieve that market penetration should be estimated. A common feature of this activity is to maximize the potential market (where frequently markets are described as being billions of dollars annually); however, it is critical to determine a realistic addressable market; this may be a sobering projected revenue, but ultimately this will determine the strategy taken in product development. Common errors are to over-estimate potential revenue and underestimate time to achieve that revenue. In general, a 510k product will generate the least amount of revenue ($2–$100 million), and the biologic/drug products will generate the largest revenues ($10–>$500 million), with the medical devices between these ($5–$200 million).
The projected return on investment (ROI) is determined from the estimated costs and timelines for product development, manufacturing, sales and marketing, and revenue generation. The ROI should be positive, and should be at a level that justifies the added risk of this investment compared with a more conservative investment of those funds. There are many assumptions, and, therefore, it is virtually impossible to be absolutely accurate in these calculations. Understanding how a product may be introduced into international markets may be a key positive additional feature to the business plan. Sound ROI assessments are critical, as they will be a major determining factor in determining whether to initially move forward on a project, or whether to continue with a project when they will inevitably be revisited at various times in the product development cycle. A common error is overly aggressive market projections. It would be unfortunate for years of effort and large amounts of funds to be wasted if the project is halted because the estimates were avoidably significantly in error. Technical hurdles, clinical challenges, and market projections are major features that can make or break a project.
Within tissue engineering and regenerative medicine, the technologies used may be relatively complex, and the resultant cost of goods may be relatively expensive. Some may not regard this as a critical issue early in the product development cycle; however, it is a very important feature, it should be considered early and addressed. The cost of manufacturing the product will be an important component of the ROI calculations. However, the manufacturing process is determined early in the product development process, and changes are difficult to make and are generally limited to small changes. Therefore, an efficient and cost-effective manufacturing process should be identified and established early in the cycle.
Bone Tissue Engineering Products and Commercialization
The applications for tissue engineering and regenerative medicine products involving bone repair include spinal fusion, fracture nonunion, maxillofacial reconstruction, and reconstruction of bone defects from trauma, tumors, infections, biochemical disorders, and abnormal skeletal development. Treatment options include autografts, allografts, materials, and, more recently, soluble mediators (e.g., BMP2 in Infuse 5 ). There have been substantial efforts made to develop new products in bone tissue engineering and regenerative medicine; however, the overall results have been limited in success. For example, OP-1 Putty for spinal fusion was found to be not approvable by an FDA review panel. 11 In contrast, Infuse has been a resounding commercial success, and has become the example for other products to emulate.
Infuse consists of a metal cage containing a collagen sponge as a carrier for BMP2. The discovery of BMP2 occurred in the early 1980s, and a very long product development effort was followed by a logical series of regulatory approvals, addressing two indications and the United States and European markets. First, FDA approval for anterior lumbar (L4-S1) spine fusion was obtained in 2002, 12 followed by European approval for treatment of acute, open tibial fractures in 2002, FDA approval for treatment of acute, open tibial fractures in 2004, 13 and European approval for anterior lumbar spinal fusion in 2005. An additional approval by the FDA was obtained as an alternative treatment to autogenous bone graft for sinus augmentations, and for localized alveolar ridge augmentation. 14
Each indication addresses a substantial clinical market where there is modest competition. A large number of high-quality scientific and clinical studies have been published [see reviews15–17 ]. The result has been substantial revenue from sales for these indications, and additional sales from “off-label” use. The dedication to the product and the concurrent investment in the R&D efforts, combined with supporting science and clinical research, lead to a remarkable revenue stream.
In contrast, efforts to commercialize BMP7 (OP-1), a related BMP family member, has not progressed as well. The target indication has been tibia nonunion fractures; however, there are several alternative (competitive) treatments (including cast bracing, bone grafting, and pulsed electromagnetic stiumulation), the market size is relatively modest, and therapeutic effectiveness has been more difficult to demonstrate. 11 The commercialization of OP-1 continues to be challenging (the product has now been sold by Stryker to Olympus who will not continue product development), and it may be that alternative additional indications will need to be approved before this product can become profitable.
The successful development of a product through to market, and addressing a clear clinical need, does not guarantee commercial success. Market acceptance is a less than scientific feature, and the clinical use of the product is dependent on a range of factors. Demonstration of therapeutic effectiveness, by performing sound clinical studies followed by presentation and publication, is a recognized method for promoting the use of the product. Other features may include reimbursement, ease of use by the surgeon, and rehabilitation and return to normal activity by the patent. These and other factors are combined into a marketing plan that plays a major role in developing the commercialization strategy for a product.
A contrast in commercial opportunities can be seen when considering potential products where there is a major clinical need but the market size is small, and, therefore, the revenue from such a product will be modest even at peak sales. The product development costs (the investment required) should still be matched to provide a positive ROI, and so this investment should also be modest. There are a number of strategies that may be considered for this, including
• Ensuring the product is regulated as a device, using a material that has already been an extensive safety record; • Using a product already in the market and seeking an additional indication for regulatory approval • Working with The Office of Orphan Products Development at FDA, which serves to advance the evaluation and development of products that are used for the diagnosis or treatment of rare diseases or conditions.
These additional restrictions add to the level of difficulty in generating an effective product; however, the awareness and the potential solutions point to possible ways forward, and exclude potential product concepts that would not be commercially viable.
Summary
The fields of tissue engineering and regenerative medicine, particularly in bone, have yielded new products that are addressing unmet clinical needs, and are beginning to demonstrate commercial success. It is likely that new products will continue to be introduced, albeit slowly. As a greater understanding of the technologies and markets grow, there is likely to be development of products with a more realistic understanding of the commercial opportunities. With sound science and business approaches working in an aligned manner, there are many opportunities for success in the short, medium, and long term.
Footnotes
Disclosure Statement
No competing financial interests exist.