
Abstract
Translation of scaffold-based bone tissue engineering (BTE) therapies to clinical use remains, bluntly, a failure. This dearth of translated tissue engineering therapies (including scaffolds) remains despite 25 years of research, research funding totaling hundreds of millions of dollars, over 12,000 papers on BTE and over 2000 papers on BTE scaffolds alone in the past 10 years (PubMed search). Enabling scaffold translation requires first an understanding of the challenges, and second, addressing the complete range of these challenges. There are the obvious technical challenges of designing, manufacturing, and functionalizing scaffolds to fill the Form, Fixation, Function, and Formation needs of bone defect repair. However, these technical solutions should be targeted to specific clinical indications (e.g., mandibular defects, spine fusion, long bone defects, etc.). Further, technical solutions should also address business challenges, including the need to obtain regulatory approval, meet specific market needs, and obtain private investment to develop products, again for specific clinical indications. Finally, these business and technical challenges present a much different model than the typical research paradigm, presenting the field with philosophical challenges in terms of publishing and funding priorities that should be addressed as well. In this article, we review in detail the technical, business, and philosophical barriers of translating scaffolds from Concept to Clinic. We argue that envisioning and engineering scaffolds as modular systems with a sliding scale of complexity offers the best path to addressing these translational challenges.
Introduction
Review papers are, in essence, a self-portrait of a field, and similar to the self-portraits of Rembrandt, should provide an honest appraisal of a field's current status, its trials, and its prospects. Our purpose in this article is to describe the current status of bone tissue scaffold engineering relative to clinical translation. As such, although we will review some technical advances in bone tissue scaffolding, our main interest is to detail the barriers to scaffold translation, how current work in the field stacks up against these barriers, and how adaptation of modular engineering may help mitigate these barriers.
We cannot characterize the barriers to, and current state of, scaffold translation, until we first define the goal of bone tissue engineering (BTE) and the role that scaffolds play in attaining this goal. The goal of BTE is to develop viable, clinically competitive approaches for bone reconstruction in Orthopedic, Spine, and CranioMaxilloFacial (CMF) surgery. Clinically competitive implies that a tissue engineering therapy provides one or more of the following: (1) improved patient outcomes, (2) reduced morbidity or complication (e.g., by eliminating the need to harvest bone graft), or (3) reduced procedural expense (e.g., through reduced operating time or patient hospitalization stay). Relative to this defined goal, BTE, and by extension BTE scaffolds, has been, to date, a failure.
What is the basis for this blunt statement? First, as extensively noted by Ratcliffe,2,3 the number of combination BTE products can be counted on one hand. These include degradable bone void fillers, which can be considered as BTE scaffolds, combined with biologics. Recombinant human bone morphogenetic protein 2 (rhBMP2) has been the most commercially successful BTE product, being utilized in up to 25% of all spinal fusion procedures, 4 and a variety of off-label applications (up to 83% of rhBMP2 use has been estimated to be off label 5 ). However, there have also been significant complications with rhBMP2 use, including patient death, dysphagia, and airway compression in cervical spine fusion,6–10 heterotopic bone formation in the spinal canal11–14 causing the U.S. Food and Drug Administration (FDA) to halt one posterior lateral fusion (PLF) clinical study, heterotopic bone formation in tibial fracture repair, 15 and retrograde ejaculation in patients undergoing anterior lumbar interbody spine fusion. 16 In addition, the FDA recently rejected the Medtronic AMPLIFY product using rhBMP2 with a composite collagen I/beta-tri-calcium phosphate (β-TCP)/hydroxyapatite (HAP) carrier for PLF product citing concerns about cancer risk in the BMP2 group versus the control group. 17 Further, there are significant questions concerning whether rhBMP2 provides better outcomes, 4 and even if so, these numbers indicate that 75% of spine fusions are still done by using traditional bone grafting methods.
For complex, large defects (especially in CMF reconstruction), the clinical gold standard remains the vascularized free-fibular graft,18–21 and indeed, no surgeon would consider using a BTE alternative, as none exist. Simply stated, there are very limited numbers of BTE clinical products, and bone grafting (auto or allo) remains the overwhelming treatment of choice. Thus, BTE has failed to achieve the goal of becoming a viable clinical alternative, despite 25 years of research, hundreds of millions of dollars of federal research funding in the United States by both the National Institutes of Health (NIH) and United States Department of Defense (DOD, including the Armed Forces Institutes of Regenerative Medicine [AFIRM]), and over 12,000 papers published on BTE in the last 10 years, including over 2000 papers on BTE scaffolds. Understanding why scaffold-based BTE has failed to date as a viable clinical alternative for bone reconstruction requires a detailed overview of the technical, business and philosophical barriers to translation.
Technical Barriers to Scaffold Translation
The first barrier to scaffold translation is addressing all the technical demands that a scaffold should fulfill to successfully reconstruct bone defects. In this sense, the technical demands are analogous to the necessary part of the necessary and sufficient conditions for a logical proof. It is necessary that technical demands be met, but meeting technical demands is far from sufficient to make a scaffold successful, as noted in the subsequent sections.
To address technical demands, we should first identify these demands and then specify quantitative targets that need to be met to satisfy these demands. This is not only good engineering practice, but also required by the FDA as a part of the Design History File and Device History File for current Good Manufacturing Practices (cGMP). Technical scaffold demands can be qualitatively summarized as the 4Fs: Form, Function, Fixation, and Formation. 22 Form is the requirement that scaffolds fill complex three-dimensional (3D) bone defects. Function is the requirement that scaffolds provide temporary mechanical load bearing within bone defects. Fixation is the requirement that scaffolds are securely attached to bone at the defect margins, eliminating motion between host bone and scaffold that could lead to nonunion and pseudarthrosis. Formation is the requirement that scaffolds enhance bone formation by providing appropriate mass transport environments, allowing perfusion, and delivering osteoinductive factors, including cells, proteins, and/or genes.
To drive engineering and material design, however (and satisfy Design History requirements for cGMP), we should define quantitative requirements associated with the 4Fs. Form is readily identified by the 3D defect geometry defined on computed tomography or magnetic resonance images. However, complex 3D defect geometry will typically have to be characterized by using 3D design data such as triangular surface facets or polylines, and require specific image processing and Computer Aided Design software. Generation of. STL data allows computational definition of defect space, specifically characterization of defect geometry in terms of volume and surface area. Function implies that the scaffold can withstand stresses under loading without failure or excess deformation. Since anatomic loading conditions are notoriously difficult to specify, scaffold mechanical design requirements have been simplified to match some fraction of native bone linear elastic properties,23–26 which have been extensively detailed.27–31 Fixation requirements are also mechanical, specifically requiring that motion at the scaffold/native bone interface be minimized32–36 (strain <2%–3% 36 ) to avoid pseudarthrosis.
Of the 4Fs, quantitative requirements governing formation are the most nebulous and difficult to specify. There are two general, yet coupled, scaffold attributes that influence bone regeneration. The first is mass transport and perfusion, which is dictated by 3D scaffold pore architecture. The second is delivery of biologics including cells, proteins, and/or genes that promote bone formation. The two are coupled, as the 3D pore architecture determines the nutrient/waste diffusion to seeded cells, provides a migration path for host cells, and determines the 3D distribution of proteins and genes and their interaction with cells as delivered from the scaffold. Mass transport can be quantitatively characterized by permeability, which relates fluid velocity in a porous medium to the pressure gradient, and diffusivity, which relates ion concentration to chemical concentration gradients. Biologic delivery is much more difficult to quantitatively define, and it depends on biologic-scaffold interactions and scaffold degradation, as well as on mass transport.
Creating a scaffold to address all the 4F quantitative requirements is an extremely challenging task, and may be implausible in some clinical applications. It is complicated by a number of factors, including (1) we don't know the target values for most of the quantitative design requirements (elastic properties, permeability, diffusivity, surface area, etc), (2) to establish design target values, we should be able to engineer scaffolds with controlled ranges of these properties to test design hypotheses, 26 and (3) these properties occur on multiple, interrelated hierarchical scales. Engineering scaffolds with controlled properties at hierarchical scales require (1) computational design methods that can represent geometry and compute effective properties (elasticity, permeability, diffusion, degradation, etc.) based on hierarchical material and pore distribution, and (2) fabrication methods that can build scaffolds with integrated biologics at multiple scales from multiple materials. It is difficult, if not impossible at the moment, for any computational design or fabrication technology to address all scales. Therefore, both the computational design and fabrication are broken into separate, but more weakly connected, pieces or modules, at different hierarchical scales. This concept of separating both design and fabrication into loosely connected modules is aptly named “modularity.” Modularity has been employed for a number of years for design and manufacturing in the computer industry, 37 but beyond this is seen as the basis of creating complex systems in technology 38 and even in nature.39–41
Hierarchical modularity in design entails computing effective properties (elasticity, permeability, diffusivity, and degradation) to meet functional and formation requirements by distribution of material and pore structures at multiple levels in 3D space. Homogenization theory42,43 has been adopted by our group44,45 and others46,47 to compute effective scaffold and biologic tissue properties directly from 3D hierarchical morphology and scaffold architecture. In addition, homogenization theories have been used within topology optimization schemes48,49 by our group50–53 and others54,55 to actually predict the optimal scaffold architecture that will give desired effective properties. Homogenization theory is an asymptotic expansion of partial differential equations that allows separation of scales. This theory assumes that a global macroscopic structure (the anatomic scaffold) is made of repeating periodic unit cells (in this case, cell refers to a mathematical unit). Thus, periodic unit cells provide a natural modularity to design in that each spatial location can be separately designed.
The controlled 3D pore architecture created by periodic unit cell design within an already complex anatomic defect 56 cannot readily be manufactured from even a single-phase material at one scale. Creating scaffolds having controlled architectures with multiple scales, multiple materials, and delivering multiple biologics (including seeded cells) is virtually impossible with a single-step fabrication process. Instead, multiple, sequential fabrication steps can be linked together to create a scaffold with multiple scales, functionalized with multiple biologics. Fabricating scaffolds with complex, anatomic shape integrated with rigorously controlled 3D pore architecture cannot be accomplished with more traditional techniques such as porogen leaching, gas foaming, electrospinning, or emulsion (see e.g., Refs.57–64 ). Solid free-form fabrication (SFF) techniques are the most viable technology for fabricating scaffolds with complex anatomic shape integrated with controlled 3D pore architecture. Indeed, since the first application of SFF to scaffolding in the late 1990s,65,66 there has been a explosion of scaffold SFF with applications ranging from cell culture to large preclinical animal models to human patients.67–104 However, SFF is typically limited in terms of feature size resolution to the hundreds of micron scale. Again, modularity comes into play when the traditional scaffold fabrication techniques can be combined with SFF to fabricate hierarchical scaffolds,100,105 or to create bioactive scaffolds with controlled architecture by linking biologics to SFF scaffolds.106–109
Modularity can be used to promote bone formation within scaffolds without negatively impacting form, function, and fixation. There is a common trade-off between the 4Fs during scaffold design, as strategies to promote formation can require manufacturing techniques or base materials that intrinsically limit scaffold physical properties that relate to form, function, and fixation. Perhaps the most extreme example are collagen-based scaffolds, which have biological properties that can be attractive for bone formation, but have elastic moduli orders of magnitude lower than native bone tissue. Modular strategies can be used to design physically robust scaffolds that promote bone formation by taking advantage of key scaffold parameters. One such parameter is pore size, as recent studies demonstrate that microporous materials can promote enhanced bone ingrowth when compared with nonmicroporous materials.110–113 Another key parameter is “osteoconductivity” provided by calcium phosphate (CaP)-based minerals. CaP materials have been widely used as bone substitutes in dentistry and orthopedics, as they bear compositional and functional similarity to human teeth and bones. 114 Common clinical examples of CaP materials include hydroxyapatite (HAP, Ca10(PO4)6(OH)2), β-TCP (β-Ca3(PO4)2), and biphasic CaP composed of HAP and β-TCP. CaP materials are osteoconductive in that they improve integration of biomaterials with surrounding bone tissue, and further serve as a template for the proper function of bone-forming cells. The favorable aspects of CaP materials have led to their extensive clinical use as bone void fillers, 115 and as coating materials on various implants to promote bone-implant bonding, 116 and a subset of studies have indicated that CaP scaffolds can also be osteoinductive at heterotopic sites in some animal models. 117 Another set of recent studies has demonstrated that ions (e.g., strontium, zinc) incorporated into Ca-based materials can induce bone formation by activating pro-osteogenic gene expression.118,119 Each of these previous studies describe bone formation parameters that can be used for scaffold design, and, importantly, these parameters can be potentially optimized without negatively impacting bulk scaffold properties.
There has been a growing demand to deliver biologically active proteins, such as growth factors, cytokines, and antibodies, which can actively guide cellular response toward new bone formation. Modularity in the form of sequential bio-functionalization procedures postfabrication can also be potentially used to deliver biologics, thus creating bioactive scaffolds. However, the majority of biologics delivery strategies do not use carriers with appropriate form, function, or fixation. The common strategy instead involves designing a biologics “carrier,” then combining the carrier with a standard orthopedic device with appropriate physical properties. Notable examples include rhBMP2-loaded or OP-1-loaded collagen matrices combined with a titanium cage120,121 or other metallic hardware. 122 Innovative biomaterials for controlled release, 123 ranging from polymer scaffolds to injectable micro- and nanoparticles, are also typically developed in a manner that requires combination with a structural device. This “combination approach” presents a challenge in clinical translation, as there is, in essence, a need to develop two separate scaffolds—one for delivery and one for appropriate form, function, and fixation. An alternative “modular approach” would involve integrating the biologics carrier into the structural device without negatively impacting physical properties. In this case, the same basic device platform can be derivatized with multiple levels of complexity, ranging from scaffold material alone to scaffold loaded with multiple biologics. One strategy to achieve scaffold modularity involves creating surface coatings. Coatings can effectively serve as carriers without significantly impacting bulk scaffold properties, and they can also be designed to impart other desirable properties (e.g., osteoconductivity, biocompatibility). In addition, it is possible to design broadly adaptable surface coatings for controllable delivery of peptides, proteins, DNA, cells, and combinations thereof. Our recent experience with nanostructured mineral coatings that bind to biologics provides one example of the utility of a modular approach.124–128 We have also developed molecular coatings that can induce bone formation,108,128–131 and the interaction between the osteoinductive molecule and the scaffold can be used to dictate the release rate. In each case, the biologics delivery is decoupled from device physical properties, thus allowing for independent optimization of each component in a modular scaffold.
It is noteworthy that combining biologics with an appropriate scaffold is just one of the several inherent challenges in biologics delivery. Whether the biologic of interest is an osteoinductive molecule, gene, or cell, there are a series of critical technical hurdles to be addressed. First, there are unique, complex dynamics in each bone defect environment that make it difficult to define a consistently desirable delivery dose and time scale. The integrity of the soft tissue envelope, status of the periosteum, and age-dependent abundance of bone-forming cell types are among the variables that are not normalized across different patient populations. Second, existing biologics carriers use very high doses delivered over relatively short timescales, which has led to well-documented side effects such as edema,4,132 heterotopic bone formation, 14 and increased cancer risk. 16 For example, the recent Medtronic Investigational Device Exemption (IDE)/investigational new drug application to deliver rhBMP2 using the composite collagen/HA/TCP matrix Amplify noted that up to 20–24 mg of rhBMP2 were needed to obtain fusion in a non-human primate model. 133 Why is there a need to deliver such a large dose of osteoinductive growth factors? One reason is likely sub-optimal growth factor release kinetics, which lead to limited activity during the bone healing process. 134 In addition, osteoinductive growth factors can induce production of their own inhibitors (e.g., BMP2-induced production of noggin), can present T- and B-cell antigens, and can influence multiple distinct cell types.135,136 Finally, a recent study indicates that more than 6500 genes are differentially regulated during bone healing, 137 which suggests a molecularly complex environment in which multiple biologics may be needed to promote optimal formation of bone and other supportive tissue types (e.g., neural, vascular tissues). However, the substantial barriers to clinically translating single biologics carriers suggest that carriers for multiple biologics are likely to encounter even greater challenges. In view of this complexity, there is a clear need to develop adaptable scaffolds that can be used to gain fundamental insights into induced bone formation in a context which can then be efficiently translated to clinical applications.
The sequential fabrication/surface modification techniques needed to create modular, adaptable scaffolds addressing all the 4Fs can be separated into parallel physical and processing modules. In this sense, very complex bioactive scaffolds can be created by modularity (illustrated in Fig. 1), mimicking the use of modularity in both man-made and natural applications ranging from aerospace technology to genetics in creating complex systems from simpler functional units.37–41 Further, since both design and fabrication in this modular framework are sequential, scaffold complexity can essentially be “dialed in,” to custom fit scaffolds for clinical indications and regulatory requirements, as discussed in the next section. Modularity also allows for separate implementation of design control and quality processes, simplifying troubleshooting and reducing expenses, which is essential to making scaffolds practical and cost effective as viable clinical approaches.
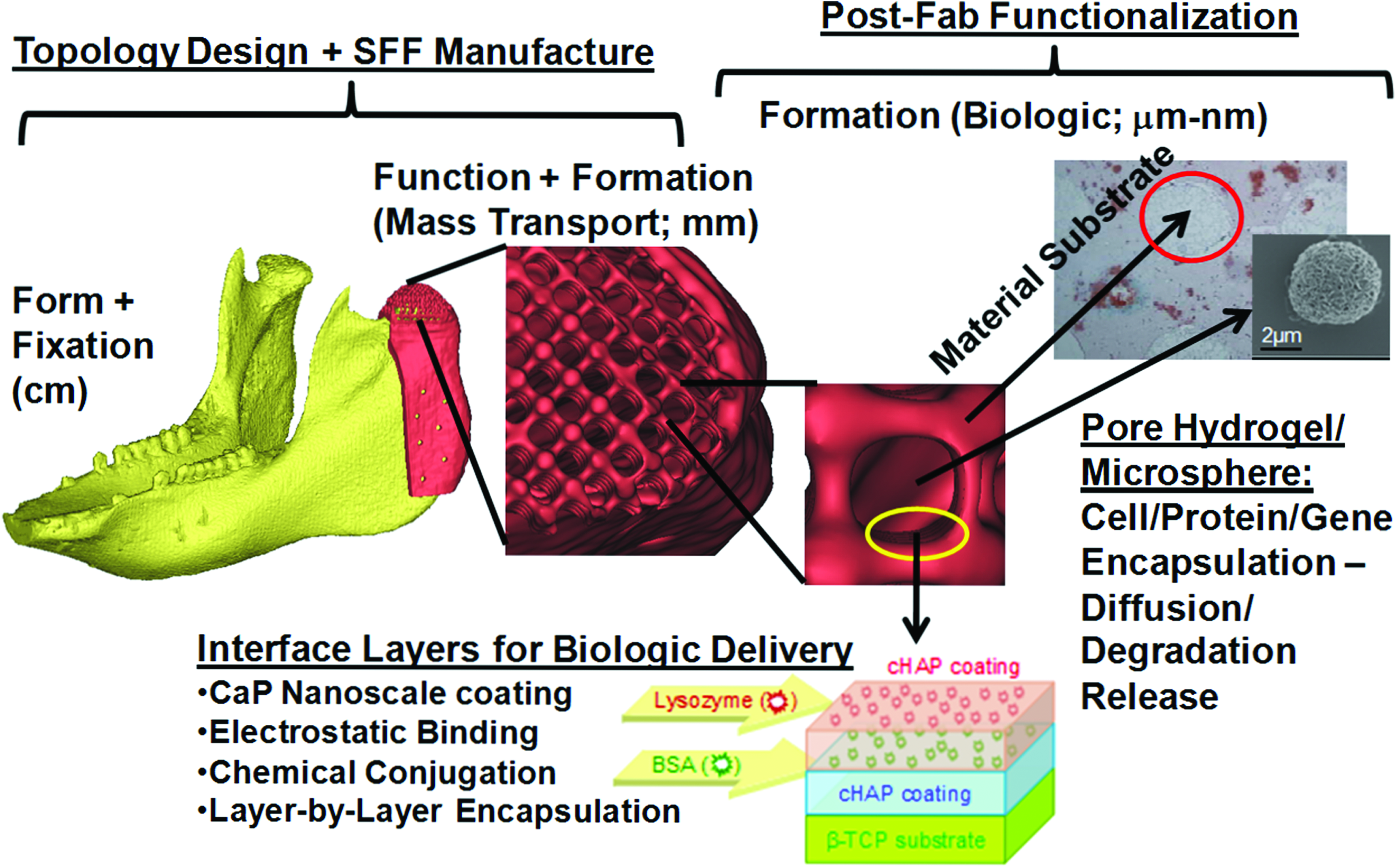
Hierarchical, modular concept for scaffold engineering. Schematic illustration of anatomic, three-dimensional porous architecture, and biologic design of a scaffold for mandibular reconstruction. This scaffold would be created through sequential engineering processes in a modular manner. CaP, calcium phosphate; SFF, solid free-form fabrication. Color images available online at www.liebertonline.com/teb
Business Barriers to Scaffold Translation
The business challenges to translation include regulatory approval, obtaining external funding for product development, obtaining physician acceptance, and, in certain circumstances, obtaining approval for insurance reimbursement. The common thread running through all these steps is the need to tailor the scaffold for specific clinical indications or needs. Although many researchers will perhaps never deal with these issues (or have a wish to), anyone interested in translating their scaffold technology must either face these issues themselves or hand off their technology in the form of a license to others who must then face these issues. Our argument is that facing these issues earlier in the research process within a modular engineering framework makes achieving downstream translation a smoother process.
The gating item to any commercialization of scaffold technology is achieving regulatory approval. Regulatory approval relevant to BTE scaffolds as devices can be broadly divided into class II (requiring 510K approval) and class III (requiring an IDE, and if successful leading to Pre-Market Approval [PMA]). Basically, class II approval will typically require that laboratory bench tests covering biocompatibility be performed in addition to relevant mechanical testing. In addition, an in vivo animal model (typically in rabbits, dogs, goats, sheep, or pigs) will be required to investigate tissue response compared with an existing clinical device (termed a predicate device) that most closely resembles the scaffold for which approval is sought. There are currently five primary clinical applications (also termed clinical indications) for BTE scaffolds that allow approval via a 510K pathway. These are (1) Bone anchoring/fixation systems, 138 (2) General Bone void filler, 139 (3) Cranioplasty (filling skull defects, a specialized case of General Bone Void Filler), (4) Dental Bone Void Filler (filling nonload bearing maxilla/mandibular bone defects, for instance, in ridge augmentation), 140 and (5) Interbody Spinal Fusion devices. 141 All these indications and the necessary tests for regulatory approval are outlined in the referenced FDA guidance documents. A common thread running through 510K bone indications is that most are minimally or nonload bearing (except for spinal fusion devices), and all indications require testing in an animal model of size equal to or larger than a rabbit with the specified bone defect. Although degradable materials such as tissue engineering scaffolds are commonly approved under general bone void filler, dental bone void filler, and cranioplasty indications, the use of degradable materials for spine fusion may face more rigorous evaluation, to the point of requiring human clinical data with the 510K submission in addition to the large preclinical animal model results.
Beyond these defined indications, or if the scaffold is used in combination with a drug or biologic such as BMP2 for any indication, an IDE will be required to test the scaffold in human clinical trials. These clinical trials are in addition to similar bench and large preclinical animal trials that are required for 510K submissions. The clinical trials are split into phases, with a phase I trial being a small trial with 10–15 patients to demonstrate safety, and a phase II clinical trial being a larger trial with 100–500 patients to demonstrate efficacy against the standard of care for the specific clinical indication. Again, as with 510K submissions, approval is given to a specific scaffold configuration for a specific clinical indication, not as a broad brush approval for multiple indications. Indeed, even though the same type I collagen sponge carrier was used to deliver BMP2 in each case, separate approval paths with a different cohort of large animal models as well as separate clinical trials were required for use in lumbar spine fusion, tibial nonunions, and sinus/ridge augmentation (Table 1).
Note that for the fourth indication, only the number of animal studies and experimental groups were provided, and the total number of animals was estimated by assuming four to eight animals per experimental group.
A number of things are readily apparent from Table 1. First, of course, is that each regulatory submission is targeted at a very specific clinical indication. Second, the number of large animals in a given preclinical study is substantial. Third, the cost of performing the preclinical studies and human clinical trials is substantial, with Ratcliffe2,3 estimating between $50 and $300 million over 8–10 years for approval of a combination product. Of course, despite the substantial investment needed, the PMA process also offers no guarantee of success. For example, a recent FDA application for use of the Medtronic AMPLIFY rhBMP2 carrier I posterolateral spine fusion included 463 patients and a 60 months follow-up, but resulted in rejection.17,133
In addition to the experimental studies required for regulatory approval of a given clinical indication, there are additional requirements to obtain marketing approval. First, in parallel with the preclinical studies required in a 510K or PMA application, biocompatibility of the base scaffold material should be established by performing bench and small animal tests as per International Standards Organization (ISO) 10993 guidelines. These tests in and of themselves may cost between $0.5 million and $3 million. Finally, approval to market a scaffold requires that cGMP to mass produce the scaffold is in place. The cGMP consists of two primary phases. The first is a Design History file, in which the scaffold design characteristics are detailed, including material, geometry, porosity, mechanical properties, and so on. In this file, the acceptable ranges for these parameters are given, in addition to the tests that will be used to determine whether the actual scaffold device meets the design criteria. The second is the Device History file, in which the history of actual manufactured scaffolds are traced, including the outcome of tests to determine whether the device meets the design specifications. If any of the manufactured devices does not meet design specifications, then it is rejected for use.
The requirements of meeting regulatory challenges, including running large preclinical animal studies, human clinical trials, and implementing cGMP, is beyond the capacity of most research laboratories, especially given the expense (>$5–$10 million for 510K; >$30–$50 million for IDE/PMA2,3), the need for dedicated manufacturing facilities and quality systems, and the need to hire dedicated personnel for regulatory, quality, and manufacturing. Further, facing the same regulatory and economic challenges, large established medical device companies are unlikely to invest the significant amounts of money necessary to bring an “early stage” scaffold technology directly from a university through to clinical approval. Their preference would be to invest in a technology where a significant amount of risk has been addressed, having made it up to or even through phase I human clinical trials.
The resulting gap between university research and high-level product development, sales, and marketing is known as the “Valley of Death.” 142 In most cases, a third entity, perhaps analogous to Charon and the river Styx, is needed to ferry the technology from the research lab to the clinic, with or without a large medical device industrial partner. This leaves the choice of a small company, likely a university spin-off, to take the technology from a university research laboratory to 510K approval (including passing 10993 biocompatability tests) and/or up to the start of phase I human clinical trials. Whether this company exists or should be created, it will need to secure outside funding to support product development. Although Small Business Innovation Research (SBIR) grants can support feasibility testing, the typical SBIR (between $0.1 and $1 million) cannot support the sustained drive toward regulatory approval of a product, especially given the current uncertainty in federal funding.
Product development by a spin off, therefore, requires support through venture capital funding. Venture capital funding, of course, seeks a return on investment (ROI) by developing a product to potential clinical application (either 510K approval or clinical trials for PMA) and then selling the spin-off company to a larger medical device company, or taking the spin-off company public. Maximizing ROI requires maximizing the potential market for a scaffold product while minimizing the risk and cost that includes regulatory approval and subsequent clinical acceptance vis-à-vis competing clinical approaches. Both aspects of ROI, maximizing market while minimizing risk, clearly necessitate defining the specific clinical indication for the scaffold. Examples include CMF reconstruction, spine fusion, and segmental long bone defects. Scaffold technologies that require extensive regulatory approval pathways of 8 years or more and greater than $100 million will likely need market value in the multiple hundreds of millions of dollars to make a venture investment worthwhile. The judgment to invest can only be made if the clinical indication is known, as this clinical indication determines the market size, the regulatory burden, and the competing therapeutic approaches.
Business challenges to scaffold translation are daunting, and it is unlikely that they can be completely addressed within an academic setting. Conversely, due to the financial risks of bringing multiple new products to the market, large device companies have a limited bandwidth for developing new products (especially those requiring a PMA such as many tissue engineering products), and are likely to rely on spin-off companies that mitigate a large share of the risk in getting scaffolds to a 510K approval or into clinical trials for a PMA. Thus, it is these small, largely spin-off companies that represent the greatest chance at translation. However, these companies rely on and collaborate heavily with academic research laboratories, and a technology cannot be simply “thrown over the wall” into the spin-off company through a license agreement. Indeed, initial venture capital investment may heavily depend on progress toward feasibility in a large preclinical animal model for a clinical indication, as more translational progress in the academic lab means a reduced risk for the venture capital investment. Further, since the spin-off company is on the clock with venture investment, the sooner it can define the clinical indications, the sooner it can begin regulatory testing and set up Quality Systems for cGMP. A common temptation for spin-off companies is to develop a breadth of technologies in several parallel applications, but this breadth can often be achieved only at the expense of depth in a focused clinical indication.
The final business challenge beyond regulatory approval and the R&D necessary for product development is physician and hospital acceptance of the scaffold product. Physician acceptance will depend on how the proposed scaffold or scaffold/biologic compares with existing clinical approaches, in most cases bone grafting techniques. Specifically, any new technology should provide better patient outcomes and/or be easier or less expensive to use in the operating room.
In summary, business barriers to translation include regulatory approval, obtaining venture funding on the order of $1 to $100 million for dedicated product R&D, and attaining physician acceptance by benchmarking against current clinical approaches. A common thread running through all these challenges is balancing risk against potential market size. Use of more complex therapies integrating scaffolds with biologics may address larger markets and more complicated defects, but also run higher risk in terms of greater costs for regulatory approval, larger R&D and quality system costs, and longer development time.
Here again, a modular approach can mitigate risks in multiple ways. First, the complexity of a modular system can be “dialed in,” adjusting its regulatory path. Consider the most complex of therapies, a combination product consisting of a scaffold made from a new degradable material directly coupled with a biologic. This combination product would face the most difficult class 3 regulatory approval (no matter what the clinical indication) in addition to satisfying the full battery of ISO 10993 biocompatiblity testing, which would include costly carcinogenic testing. The ISO 10993 tests alone would likely cost $3–$5 million and take 1–2 years to complete. Another 6–8 years and likely a $50–$100 million investment would be required to perform the large preclinical animal testing and human clinical trials for one indication. This path would likely be complicated by the fact that the ISO 10993 tests would have to be completed before IRB approval to begin human clinical trials. This path, of course, is not only an arduous regulatory path, but also presents significant risk to potential investors. If, however, the technology is modular, then the scaffold technology could be developed separately from the biologic delivery. For example, the scaffold technology alone could be initially developed as a bone void filler, with biocompatibility tests and preclinical tests completed to achieve 510K approval. A product with potential for at least modest revenue would be in place, biocompatibility tests and some preclinical results achieved, to establish regulatory history. Further, the quality systems necessary to engineer the scaffold would be in place as required by the FDA for regulatory approval. Regulatory complexity of combining the scaffold with the biologic would be reduced, as biocompatibility tests and some preclinical data would be on record. Thus, the additional regulatory complexity would involve studying the combination therapy in a large preclinical model and in human clinical trials. Second, although additional Quality Systems covering a biologic attachment would be required, Quality Systems governing the design and manufacturing would be in place. This would again reduce the investment risk to extend the scaffold product to a combination product. Third, a modular scaffold would provide separation of processing steps, thus reducing investment and maintenance costs as troubleshooting may be done on isolated modules, which is a significant advantage of engineering systems using modular approaches. 37 In summary, a modular approach beginning with a scaffold alone or with surface modified scaffold may achieve approval under a 510K pathway, followed by a scaffold/biologic combination that would require a PMA. Approval of the scaffold alone would provide a history of biocompatibility that may aid additional regulatory applications.
Philosophical Barriers to Scaffold Translation
Addressing the technical and business challenges of scaffold translation requires significant resources and a focus on specific clinical indications. Whether conscious or not, translational research targets and the allocation of resources to meet these targets reflect an underlying philosophy or model. Two fundamental scientific research models, termed the “linear” and “quadrant” models, were proposed by Donald Stokes in his book “Pasteur's Quadrant: Basic Science and Technological Innovation.” 143 The linear model (often ascribed to Vannevar Bush, science advisor to Presidents Roosevelt and Truman, and father of the National Science Foundation) proposes a separation of basic and applied research, where basic research is done without regard to final application, and applied research is done after basic research, piecing together basic research results to address a problem. The quadrant approach integrates basic and applied research, with both motivated by a specific problem, and the drive to solve the problem motivating both applied (near term) research, and basic research to investigate fundamental phenomena relative to the defined problem.
How do such models relate to translation of BTE scaffolds? Implementation of the linear model would proceed from initial development of a material, to in vitro testing of osteoprogenitor cells on the material, to small animal (mouse and rat) testing, to large animal testing for specific clinical indications against competing clinical approaches (e.g., long bone or CMF defects in sheep, pigs, goats, or non-human primates) to clinical testing. As one proceeds, it would become necessary to create more complex scaffold systems, for example, to withstand the functional loads of preclinical or clinical models, clearly not a necessity in early cell culture or small animal testing stages. Clinical indications would not be typically specified until the preclinical large animal model. In contrast, implementation of the quadrant model would proceed by first defining a specific clinical problem (e.g., long bone defect, CMF defect, or spine fusion), and then developing a modular scaffold system with defined components (3D material architecture, osteoconductive interface, and delivered biologics) to test first in the specific preclinical defect. Difficulties in the large animal system may lead to further testing and/or screening of basic phenomena in cell culture or small animal models.
What model prevails in BTE scaffold research and BTE in general? The closest, as evidenced both by resource allocation (funded grants) and research targets (percentage of published papers), is the linear model. However, if the classic Bush linear model as defined by Stokes was followed, then one would expect the number of publications on large preclinical animal work for specific defects to increase, and that funding targeted at specific clinical indications would also be on the rise. However, examination of both recent funding and publications indicate that this is not the case. A review of specific aims and publications from 40 grants funded by the NIH for BTE (as per the NIH website: http://projectreporter.nih.gov/reporter.cfm; Table 2) in 2010 reveals that only three grants (7.5%) had aims that utilized animal models of rabbits or larger and/or had published papers by using animal models larger than rats (rabbits, dogs, sheep, pigs, or goats). Estimated funding for these aims totaled $0.45 million of a total $17.5 million (2.6%).
Dollar amounts and grants were obtained from the National Institutes of Health website: http://projectreporter.nih.gov/reporter.cfm. Animal models were determined from the Specific Aims.
Funding for grants in which aims were split into small animal/cell culture, and large animals were double counted as it was not possible to split costs by aim.
Indicates that an animal model was proposed, but as of the paper submission, no publication was on PubMed.
Beyond the US federal grant support and private investment, there are in addition, of course, numerous other national funding agencies in Asia and Europe, including combined European Union funding for translational research. The European Union has in this sense pursued a more coordinated effort in translational research under its Framework Programme (FP) initiatives. 144 The recently completed FP5 initiative funded €66 million (∼$93 million) toward translational research in biomaterials related to tissue engineering from 2004 to 2007. Within this funding, €7.3 million ($10.4 million) was used to establish a virtual European Tissue Engineering Center focused on bone and cartilage engineering and €2.3 million ($3.3 million) on an artificial bone graft project. This is roughly an average €3.2 million/year ($4.5 million/year). Although preclinical animal testing was stated as an outcome of both sub-projects, the reference did not specify the amount of funds dedicated to large animal studies. However, given that numerous other topics were investigated, the amount of funding dedicated to large preclinical models is likely to be on the order of, or less than, NIH funding, both in terms of absolute funding (likely <$0.5 million/year) and percentage of funds.
The final source of translational research funding outside of public government funding and private investment are nonprofit foundations. For example, the Wallace H. Coulter Foundation specifically has a mission of funding translational research in biomedical engineering that requires a clinician/researcher investigator team, a definition of clinical markets, and intellectual property before beginning the research project. In addition, the AO Foundation is another source of translational research funding, with a particular focus on musculoskeletal conditions. However, although foundation funding is a good jumpstart to translational research, funding is by design and necessity (as Foundations like Coulter fund a large range of biomedical engineering translational projects and do not focus on tissue engineering) still aimed at the early research stages, with funds limited to $0.1 to $1 million per project, on the order of a phase II SBIR grant. This level of funding cannot sustain the $10–$100's millions necessary per product to obtain regulatory approval.2,3 Instead, such funding levels typically require a consortium of private investors.
Funding allocation is essentially an “input metric” that illustrates the motivation for translational research and development through monetary support. An intermediate “output metric” is peer-reviewed publication. A PubMed search on the keywords “bone tissue engineering” (BTE) up to November 2010 revealed a total of 12,219 publications, whereas a search on “BTE scaffolds” revealed a total of 2268 publications. Of the “BTE” references, only 523 utilized animals larger than a rat (4.2%), whereas of the “BTE scaffold” references, only 130 utilized animals larger than a rat (5.7%). If one further categorizes these animal model studies into large preclinical models for specific clinical indications that present challenges to bone reconstruction (mandibular defects (17),97,145–160 long bone defects (14),161–174 cranial defects (7),175–181 and spine fusion (14)67,182–194) and utilize BTE in some form (degradable scaffolds with/without biologics), there have been ∼52 studies accessible by PubMed up to 2010. Of these studies, four have acknowledged NIH support (7.7%), whereas none (0%) acknowledged DOD support (including AFIRM, which lists significant craniofacial and long bone reconstruction as two of its clinical outcome goals). In comparison, six studies acknowledged support from local or national funding agencies in China (11.5%), four studies acknowledged support from local or national funding agencies in Germany (7.7%), two studies acknowledged support from local or national funding agencies each (3.9%) in France, Taiwan, and Singapore, and one study each (1.9%) acknowledged support from funding agencies in Switzerland, Sweden, Italy, Japan, and the Netherlands.
Thus, no matter what metric is applied for either resource allocation or research targets, research on specific clinical indications using animal models sufficient to attain regulatory approval (510K or PMA) amounted to between 2% and 8% (except for China, with acknowledged funding of over 11.5% of large preclinical studies) of total funding and publications. Indeed, summing the total number of animals used in the peer reviewed published large preclinical animal studies shows that a total of 149 animals were used for long bong defects, 64 animals for cranial defects, 116 animals for mandibular defects, and 202 animals for spine fusion. As a comparison, one PMA study on BMP2 for posteriolateral lumbar spine fusion used 95 large animals, whereas another PMA study on BMP2 use for tibial nonunions used 112 large animals (Table 1). Thus, one FDA study for one clinical product used the equivalent of 47% to 75% of all the large animals published in BTE research for the same clinical indication for the past 10 years, of which only 8% were acknowledged and funded by NIH and 0% by the DOD (including AFIRM, which invested $81 million specifically for clinical translation) in the United States.
These results clearly indicate that despite the hand wringing over translation, resource allocation (i.e., grant funding) and research emphasis (i.e., publications) are not geared toward translation. An apt analogy is having an Olympic running team where the stated goal is to win gold medals in sprints and the marathon. If the only practice time is spent in running sprints, then no matter how loudly the coach proclaims that the goal is to win gold medals in both events, clearly no gold medals will be won in the marathon. Similarly, no matter how loudly the field proclaims that translation is the goal, if funding agencies, study sections, and journals favor scaffold studies in cell culture and small animal models, but not pre-clinical models targeting relevant bone defects or clinical trials (as reflected by the roughly 5% resource allocation for clinically relevant defect studies), then translation will simply not occur. In looking at the data regarding translation, it is clear that the field is not “putting our money where our mouth is.”
Clearly, the current path has inhibited rather than enhanced scaffold translation. Although there is a clear and necessary role for discovery-based basic science in tissue engineering, there should be a balance between basic science and translational research and development, which is not achieved when well more than 90%–95% of resources are allocated toward discovery research and the rest toward translation. Here, we define translation as engineering a scaffold for a specific clinical indication and testing that scaffold in a large in vivo model which realistically stimulates the target human clinical indication. Although some may argue that it is not the purpose of federal funding to support translation, translation will not realistically occur without significant federal support. Others may argue that federal funds should focus on fostering innovation in the form of discovery research. However, innovative discoveries are of limited value if they cannot be used in clinical applications, and it is clear that substantial innovation is needed to overcome the hurdles to clinical translation. Given the significant business barriers, chances of translation increase when technology coming out of academic laboratories is more mature, can readily be implemented in quality controlled scalable processes, and realistically addresses important specific and, perhaps, multiple clinical indications. In this path, modularity that supports platform technology production is a significant benefit.
The question becomes as to what can be done to improve federal support of translation. In the current economic climate, it is unlikely that NIH or other federal funding sources will increase. Therefore, a conscious decision should be made to shift a portion of funding to translation focused projects. Specifically, NIH or other federal agencies should set aside research money for specific BTE translational projects. This percentage will likely need to increase to 25%–30% of BTE funding, compared with the current 2%–8% levels, if increased translation is to be a reality. Increased federal funding of targeted large preclinical animal models would encourage private venture funding by reducing regulatory and market related risk. Specifically, risk would be reduced by successful demonstration of clinically relevant scaffold therapies against clinical gold standards in an academic setting. Such results would demonstrate the capability of performing necessary preclinical studies as well as demonstrating that a new technology could compete against a clinical standard, allowing assessment by, and attracting interest of, venture funding and large medical device companies.
Projects funded under a translational emphasis, however, should be clearly defined and more rigorously thought out than in previous funding efforts. We would suggest that such projects target a specific clinical indication, and further, layout out a regulatory approval path, quality systems implementation of the proposed technology, and the potential market in the proposal. The proposal should delineate where the proposed work fits in the path. In addition, such proposals should be multidisciplinary, specifically including clinicians who practice in the clinical indication, and who would help implement large preclinical animal model testing, another requirement of the project. Some components, such as detailed regulatory insights and marketing insight/data, may be difficult to build into translational research proposals in their formative stages, due to the need for highly specialized and costly expertise. The need for such unique resources in all translational research endeavors perhaps presents an opportunity for funding agencies or institutions to create core services that provide the needed expertise.
To help direct such targeted funding, significant input is needed not only from the research community, but also from the clinical community, the venture capital community, and the corporate medical device community. Having dedicated workshops where practitioners from each community come together to draft the requirements for an RFA is necessary. Specifically, having both clinicians and industrial partners from CMF, orthopedics, and spine surgery will be critical.
In conclusion, there are significant barriers that explain the dearth of scaffold translation. These include the numerous technical and business barriers (especially definition of and engineering scaffolds for specific clinical indications) that present an engineering paradox. Technical barriers to fulfill the 4Fs push increased scaffold complexity, but business barriers penalize complexity in terms of increased regulatory and quality systems cost and implementation time. These difficulties may be mitigated through engineering modularity, which allows for a sliding scale of complexity. Finally, however, there are philosophical barriers that cannot be adequately addressed by individual labs or product development teams, but only by the field as a whole if translation is to be increased. These include, first, recognizing the special needs of translational science and engineering and, second, the need to allocate sufficient resources (both funding and intellectual recognition in terms of publishing) to these needs.
Footnotes
Acknowledgments
S.J.H. would like to acknowledge current and previous funding from the NIH supporting research cited (AR 053379, DE 016129; DE 13608) and the Coulter Foundation. WLM would like to acknowledge current and previous funding from the NIH (AR052893, AR059916), the Coulter Foundation, and the AO Research Foundation (F-07-65M and Exploratory Research Grant) supporting the research cited.
Disclosure Statement
The authors are co-founders and shareholders in a company, Tissue Regeneration Systems Inc, which develops scaffold technology.