
Abstract
The treatment of degeneration and injury of articular cartilage has been very challenging for scientists and surgeons. As an avascular and hypocellular tissue, cartilage has a very limited capacity for self-repair. Chondrocytes are the only cell type in cartilage, in which they are surrounded by the extracellular matrix that they secrete and assemble. Autologous chondrocyte implantation for cartilage defects has achieved good results, but the limited resources and complexity of the procedure have hindered wider application. Stem cells form an alternative to chondrocytes as a source of chondrogenic cells due to their ability to proliferate extensively while retaining the potential for differentiation. Adult stem cells such as mesenchymal stem cells have been differentiated into chondrocytes, but the limitations in their proliferative ability and the heterogeneous cell population hinder their adoption as a prime alternative source for generating chondrocytes. Human embryonic stem cells (hESCs) are attractive as candidates for cell replacement therapy because of their unlimited self-renewal and ability for differentiation into mesodermal derivatives as well as other lineages. In this review, we focus on current protocols for chondrogenic differentiation of ESCs, in particular the chemically defined culture system developed in our lab that could potentially be adapted for clinical application.
Introduction
T
Many surgical procedures have been developed to help restore damaged articular cartilage, such as arthroscopic lavage and debridement,1–4 bone marrow stimulation, including abrasion arthroplasty,5–8 drilling, and microfracture.2,9–11 These techniques have some benefits in generating fibrocartilage or hyaline-like cartilage, but this can lack the mechanical durability needed in load-bearing joints.3,12
Osteoarthritis (OA) is the most prevalent disease that compromises the function of articular joints and damages cartilage. OA increases with age and affects 10% of men and 18% of women older than 45. The OA-affected joints are painful, stiff, and often deformed, and advanced disease causes severe physical handicap and immobility. Pain relief provides some symptomatic treatment without any benefit in delaying progression or reversing the degeneration of the joint. OA is, thus, a major social and healthcare burden. 13
Since cartilage is produced and assembled by chondrocytes, the only cell type in cartilage, a feasible way to regenerate cartilage is to ‘re-grow’ it with a fresh supply of chondrocytes. This has been achieved by autologous chondrocytes implantation (ACI), which has been applied since 1994 with some success and uses the patients' own chondrocytes expanded in culture.14–23 However, the long-term outcome of ACI is not significantly different from that of microfracture, with some clinical improvement for both approaches but little justification for the large additional cost of ACI.24,25 A particular difficulty is the limited amount of nondisease-affected cartilage available for obtaining appropriate cells, and this is an additional challenge for treating older patients who might need cartilage repair. Stem cells, especially adult MSCs, overcome the resource limitation of primary chondrocytes, as they grow well in culture and they also have good long-term safety without risk of tumor formation. 26 However, adult stem cells have drawbacks, as their ultimate proliferative capacity is limited and they show variable potential for differentiation. As an alternative, pluripotent cells (embryonic or induced) are much more attractive because of their unlimited self-renewal capacity and ability to differentiate into a wide range of somatic cell lineages.27,28
The use of human pluripotent stem cells (PSCs) for cartilage repair has been investigated only more recently and much less than the use of adult stem cells. So far, the size, homogeneity, and stability of the cartilage formed have been insufficient for any clinical application. Furthermore, most reported protocols depend on animal products in the medium with risks of xenobiotic transfer, preventing clinical translation. This review will focus on recently developed protocols for generation of chondrogenic cells from human embryonic stem cells (hESCs) and the potential of induced pluripotent cells for human cartilage repair.
Development of Human Articular Cartilage
In early embryonic development, limb buds are formed from paraxial and lateral plate mesoderm emerging from the posterior region of the primitive streak at gastrulation. 29 In humans, they appear around day 26 for the upper limb and day 28 for the lower limb. Wnt3a and activin/nodal signaling pathways are involved in both the formation of the primitive streak and the differentiation of epiblast cells and hESCs into a proposed bi-potent mesendoderm population.30–32 Mesodermal differentiation is controlled by the bone morphogenetic protein family (BMP; especially BMP4 and BMP2) and fibroblast growth factor (FGF) families.33–36 The paddle-shaped limb bud consists of a mesenchymal core surrounded by ectoderm on all but its proximal side. The distal border of the ectoderm expands to form the apical ectodermal ridge (AER).
Once matured, the AER is maintained as a transient structure for about 2–3 days, while the mesenchymal skeletal progenitors continue to proliferate and differentiate forming a fully patterned limb. Cartilage development in the limb rudiment starts with the formation of the cartilage anlage, a condensed population of undifferentiated mesenchymal cells, which have migrated into the limb field. 37 The MSCs in the condensed population differentiate into chondroprogenitors. Their fibroblast-like morphology changes to a spherical cell shape and they begin secreting an ECM that is enriched with type II collagen and proteoglycans, such as aggrecan. Chondrogenesis initiates from the central area of the mesenchymal condensation and then radiates outward. After becoming surrounded by ECM, chondroblasts lose cell–cell contact, become chondrocytes, and organize into zones that form growth plates. The chondrocytes progress through an ordered process of proliferation, maturation, and hypertrophy with calcification within the growth plates. A separate zone of chondrocytes at the ends of rudiments segregates early in development to form the articular chondrocytes, which produce and maintain articular cartilage and remain as chondrocytes throughout life. The articular cartilage ECM is mainly composed of type II collagen and glycosaminoglycans. However, the composition varies spatially from the superficial to the deep zone, resulting in graded mechanical properties. 38 In the early development of chondrocytes, the master transcription factor driving chondrogenesis is SOX9, a member of the high-mobility-group domain transcription factor family. 39 SOX9 associates with SOX5 (L-SOX5) and SOX6, 40 to control chondrogenic differentiation, maintain the chondrocyte phenotype, and directly regulate expression of ECM molecules. 41 SOX9 is expressed in chondroprogenitor cells and throughout the life of chondrocytes, but its expression is turned off during growth plate chondrocyte hypertrophy. It positively regulates the transcription of Col2a1 by binding to the promoter region42,43 with coactivators including Znf219, 44 PGC-1α, 45 and p300.46–49 RUNX2 is the key transcription factor controlling bone formation and is also involved in the early stages of chondrocyte condensation as well as osteoblastic differentiation of MSCs. 50 The expression of these two transcription factors, SOX9 and RUNX2, directs key stages in the differentiation of MSCs, and their expression overlaps both temporally and spatially during embryogenesis. Since chondrogenesis is initiated, SOX9 induces degradation of RUNX2 by directing it to the lysosome for breakdown while during osteoblastic differentiation RUNX2, inhibits the transactivity of SOX9 and blocks chondrogenesis. 51
Signaling molecules secreted by the AER and progress zone modulate chondrogenesis, and are crucial regulators of skeletal development. These include members of the transforming growth factor (TGF)-β superfamily, such as BMPs, growth and differentiation factor-5 and TGF-β1, 2 and 3, Wnt family ligands (WNT3a, 5a and 7a), sonic hedgehog and Indian hedgehog, and also FGF family (FGF2,4,8,10 and 18)52,53 members. Retinoic acid (RA), which is the most active metabolite of vitamin A, profoundly regulates the development of multiple organs, but is particularly important in skeletal development. RA inhibits the transactivity of SOX9 54 but induces the expression of RUNX2. 55 RA clearly has a dual role in chondrogenesis: It controls the timing of condensation and chondroblast differentiation early on, and coordinates the maturation and replacement by bone at later stages. During long bone development in mammals, the original cartilaginous template is replaced by bone through endochondral bone formation. This involves growth plate chondrocyte hypertrophy, matrix mineralization, blood vessel invasion, osteoblastic differentiation, and calcified cartilage matrix remodeling and removal. Although articular cartilage forms a thin calcified layer at its junction with subchondral bone, it is highly resistant to vascular invasion and factors such as endostatin are involved in the maintenance of its avascularity. 56 Other factors, such as Notch signalling molecules, 56 BMP family growth factors, 57 and transcription factor C-1-1, 58 also play roles in regulating the development of articular cartilage. So, the formation and maintenance of articular cartilage is controlled by the cooperative activity of a network of signaling mechanisms, which include key transcription factors, extracellular signaling molecules, and interaction with ECM (Fig. 1).
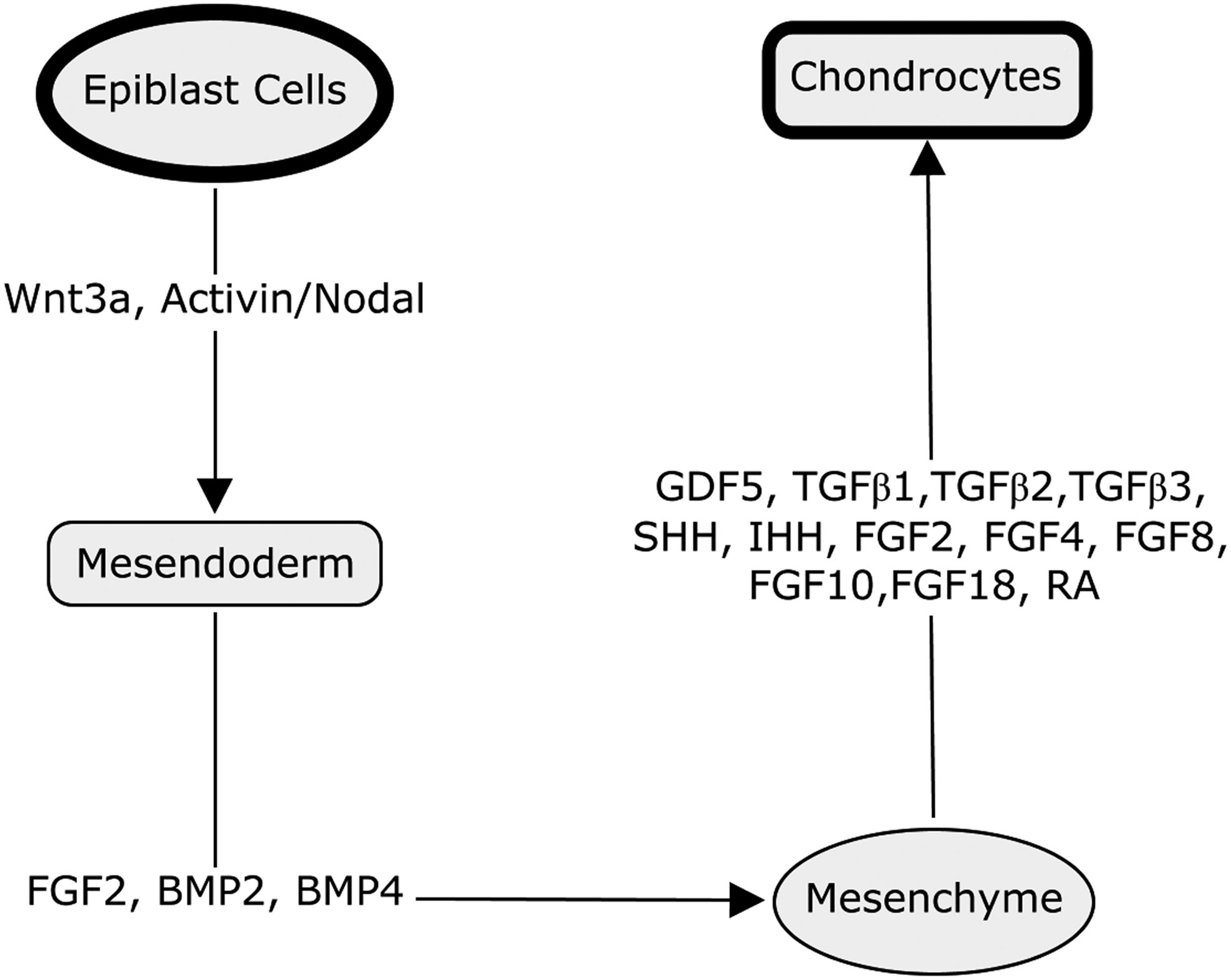
Schematic diagram indicating some of the key factors that play roles in the differentiation of mammalian pluripotent epiblast cells progressively though a sequence of stages to generate chondrocytes.
PSCs and Their Applications
The pluripotent cells from the inner cell mass of mouse blastocyst-stage embryos were first established as cell lines in culture by Martin Evans and Matthew Kaufman in the United Kingdom 59 and Gail Martin in the United States. 60 The term ‘ES cell’ was introduced to distinguish these embryo-derived pluripotent cells from teratocarcinoma-derived pluripotent embryonal carcinoma (EC) cells. 60 In 1998, the first hESC lines derived and established from human blastocysts were reported. 27 In 2006, a further breakthrough occurred, this time in cell reprogramming; induced PSCs (iPSCs) were generated from mouse embryonic fibroblasts (MEFs) by introducing four genes, Oct3/4, Sox2, c-Myc and Klf4. 61 This was quickly followed by reports that human somatic cells could also be reprogrammed into PSCs using the same factors 62 or by using NANOG and LIN28 instead of c-MYC and KLF4. 63 This process has since been replicated in similar and related ways by many groups across the world. The reprogrammed cells (iPSCs) show many of the characteristics of hESCs, as they express OCT4, NANOG, SSEA-4, TRA-1-60, TRA-181, alkaline phosphatase, and a high level of telomerase. The beauty of these PSCs is that they share with ES cells the ability to maintain prolonged undifferentiated proliferation with stable developmental potential to form derivatives of all three embryonic germ layers, even after long-term culture. In addition, no embryo is required for their derivation. Further developments, including gene transfer without c-Myc, reduces the risk of tumor development from iPSCs, which was observed in murine iPS-embryo chimeras in early studies 64 and in recent years, reprogramming has been achieved using nonintegrating vectors, 65 thus avoiding the risk of tumorigenic insertional mutations. There are still a number of problems to be overcome with iPS cells, not least the low efficiency of generation and epigenetic differences (with epigenetic memory of cell of origin) and possibly acquired mutations.
Pluripotent cells provide extensive self-renewal with the potential for scale up as well as the plasticity of multilineage differentiation capability.27,66 The development of iPSCs offers a further advantage for potential clinical application, as it enables the harvesting of cells from a patient to derive iPSCs carrying the identical genetic mutations(s) of the patient. This is likely to facilitate drug development in the near future, using human differentiated disease models produced from these cells. Patient iPSCs should also avoid immune rejection if the cells are used for autologous therapy (after gene correction).61–63 Murine syngeneic iPSC-derived teratomas were rejected because of carryover of embryonic immunogenic antigens, 67 so further research is required before such cells can be considered suitable for therapy although transplantation of primate iPSC-derived neural progenitors indicated more promising survival. 68
PSC-Based Strategies for Cartilage Repair
Generation of chondrocytes from pluripotent cells through embryoid bodies
Embryoid bodies (EB) were first described as the cystic structures formed by teratocarcinoma-derived EC cells. The three-dimentional (3D) structure of the EB is similar to the early postimplantation embryo, and the cells in EBs are able to differentiate into the three germ layers in serum containing medium.69,70 EBs have been used as a model to study embryo development and to validate ESCs differentiation.70–79 Since it was thought that key signaling occurs within the 3D EB, inducing more efficient early differentiation than culture in two dimensions, EBs have also been used as a starting point for targeting differentiation to particular cell phenotypes. Differentiation of mouse ES cells in vitro into chondrocytes via EBs was modulated by members of the TGF-β family (TGF-β1, BMP-2, and BMP-4). The application of TGF-β1 was found to decrease chondrogenesis slightly, whereas BMP-2 or 4 induced chondrogenic differentiation. The function of BMP-2 on chondrogenesis of ES cells was found to be dependent of the time of application; it induced chondrogenic differentiation only when applied during EB development from day 2 to 5. 80 Chondrocytes, isolated from spontaneously differentiated EBs, initially appeared to de-differentiate in culture but later re-expressed the characteristics of mature chondrocytes. Surprisingly, TGF-β3, which is able to promote chondrogenic differentiation in cultures of human MSCs, 81 completely blocked the process of re-differentiation. It has also been shown that chondrocytes isolated from murine EBs retain some plasticity at this stage and can transdifferentiate into other mesenchymal lineages, such as osteogenic and adipogenic cells. 82 Cartilage tissue can be formed from cells in murine EBs by application of growth factors (TGF-β3 and platelet-derived growth factor-BB [PDGF-BB]) direct, or to cultures of the disrupted EBs as a micromass or pelleted cell mass, mimicking formats used for MSC-derived chondrogenesis and cartilage formation.83–85 Human dissociated EBs have been differentiated into chondrocytes in a monolayer culture by application of 1 ng/mL TGF-β1, 5 ng/mL FGF2, and 10 ng/mL PDGF-BB, while the best combination for the pellet culture was determined to be 100 ng/mL BMP7 and 10 ng/mL TGF-β1. 86 These hESC-derived chondrocytes have been shown to form a hyaline-like neocartilage layer when implanted in a hyaluronic acid (HA) hydrogel in a rat osteochondral defect.86–88 Indeed iPSC-derived EBs have also recently been generated from osteoarthritic patients and differentiated toward the chondrogenic lineage although the differentiated cells were not sufficiently characterized to be confident of outcome. 89
Generation of chondrocytes from ESCs by co-culture or conditioned culture
Another strategy used to direct differentiation to different lineages is the co-culture of embryonic and other stem/progenitor cells with fully differentiated chondrocytes. Some success in the chondrogenic differentiation of hESCs was achieved by indirect co-culture with mature chondrocytes using inserts carrying a porous membrane, which allowed the diffusion of signaling molecule produced by the chondrocytes to hESCs. The hESC-derived chondrocytes were shown to produce a cartilaginous ECM. 90 In another study, Flk-1-positive predifferentiated cells from murine EBs co-cultured with porcine articular chondrocytes, formed cartilage tissue in nude mice 4 weeks after implantation. 91 The Transwell™ system, used to generate 3D cartilage-like matrix in vitro from MSCs, 92 has also been adapted to co-culture hESCs with chondrocytes. This method does not generate EBs as intermediates, but drives the chondrogenic differentiation of hESCs, which were able to form cartilage tissue both in vitro and in vivo. 93 However, the culture system is complicated and efficiency is low.
Generation of chondrocytes from ESC- or iPSC-derived mesenchymal cells
Human MSCs isolated from adult bone marrow have the potential to differentiate into mesodermal tissue lineages, including bone, cartilage, fat, tendon, muscle, and marrow stroma and may have some other lineage potential.
94
However, there are limitations with the use of MSCs for applications requiring a large number of cells. Expansion changes the phenotype of MSCs and can result in spontaneous transformation.
95
In addition, MSC numbers, proliferative ability, and differentiation capacity decline with age.94,96 To overcome these difficulties, it is possible to use pluripotent ESCs to derive MSCs for possible applications in tissue engineering and cell therapy. HESCs have been differentiated to MSCs using the ‘Raclure method’, which mainly employed the cells at the edges of the hESC colonies for differentiation.
97
Indeed, clinically compliant MSCs have been derived from CD105+, CD24− differentiated cells isolated from hESCs. These cells share a similar phenotype to bone marrow (BM)-MSCs and can differentiate into cartilage, bone, and fat. Compared with the BM-MSCs, these hESC-MSCs have a substantial proliferative capacity in vitro and were reported to be able to undergo at least 35 population doublings while maintaining a normal diploid karyotype with a stable gene expression and surface antigen profile.
98
hESC-MSCs were also obtained by culturing the hESCs in MSC-medium (Dulbecco's-modified Eagle's medium supplemented with 10% fetal bovine serum, 2 mM
Feeder/serum free culture of ESCs and generation of chondrocytes
The standard form of culture and expansion of hESCs has relied on the use of irradiated/proliferation-deficient mouse feeder layers of fibroblast origin. 27 However, since this method exposes cells to xeno-antigens from the feeder cells and animal cell products, this precludes its use for any clinical application (Fig. 2). 105 Furthermore, the conditions are undefined and suffer from batch-to-batch variation of serum. The use of feeder cells has been avoided by culturing hESCs on Matrigel (rodent basement membrane tumour extract) or on, for example, laminin, in medium conditioned by MEFs. Under these conditions, hESCs maintained pluripotency in vitro and were still able to form teratomas in severe combined immunodeficiency mice. 106 A number of other groups have also developed methods for feeder-free hESC culture incorporating different substrates and avoiding animal serum or conditioned medium.107–110 More recently, Chen et al. have reported a simplified minimal component defined medium for PSC culture. 111 In addition, a serum-free hESC culture system was developed by Baxter et al. using FGF2, activin A, and neurotrophin 4 with fibronectin as culture substrate. 112 Culturing hESCs under these conditions without either animal feeder cells or products will promote the clinical application of hESC- based cell therapies.

The different pathways that have been exploited to differentiate human embryonic stem cells (hESCs) toward chondrocytes.
As a step toward this end, feeder-free cultured hESCs, but maintained in MEF-conditioned medium, have been shown to generate chondrocytes by co-culture with primary chondrocytes using the Transwell™ system. 93 However, this still relied on animal serum in the MEF-conditioned medium. To facilitate clinical application, a defined hESC differentiation protocol for generation of chondrocytes was developed in our lab. 113 This protocol is based on the sequential signaling pathways active in embryonic development and outlined earlier in this review. The sequential changes in growth factors drive the differentiation of hESCs through primitive streak-mesendoderm and mesoderm intermediates to a chondrogenic phenotype (Fig. 3). The protocol exploits the feeder-free, serum-free, chemically defined hESC culture medium previously developed 112 and utilizes matrix-coated substrates. In addition to the advantage of feeder-free and serum-free culture, this short, 14 day protocol, gives an 8.5-fold expansion of the cell population as they differentiate to SOX9-positive chondrogenic cells. Importantly we obtained between 75% and 97% SOX9-positive cells with four different hESC cell lines (three out of four lines gave 95%–97%), suggesting a much higher efficiency than in most hESC chondrogenic differentiation protocols (Table 1). Indeed, the protocol has now been applied to human iPSCs with a similar level of success, giving 96%–97% SOX9-positive cells with the two iPSC lines tested (Cheng et al. unpublished), and the induced cells showed high expression of collagen II as well as the chondroitin sulphate proteoglycan, aggrecan. This suggests that the heterogenity of pluripotent cell chondrogenesis in our protocol is much less than suggested for MSCs. 114 However, we observed some cell death at the transition period from ESCs to mesendoderm in our protocol, which indicates that some ESCs could not respond to the growth factor milieu and thus enter a death pathway. This confirms that there is a degree of heterogenity in cultured hESCs as described for Nanog 115 and some signaling pathways, such as BMP, Nodal, and FGF. 116 Importantly, there was no evidence of pluripotent cells remaining at the end of the protocol. These promising results suggest that the protocol efficiently drives human chondrogenesis and could provide a suitable method for generating chondrocytes of the required standard for clinical application. We have tested the ability of the generated cells to form cartilage in vivo using an osteochondral defect model in nude rats. We have found that these cells can form hyaline cartilage, identified at 4 weeks and further developed by 13 weeks after implantation of the cells, without evidence of any aberrant tissue formation.
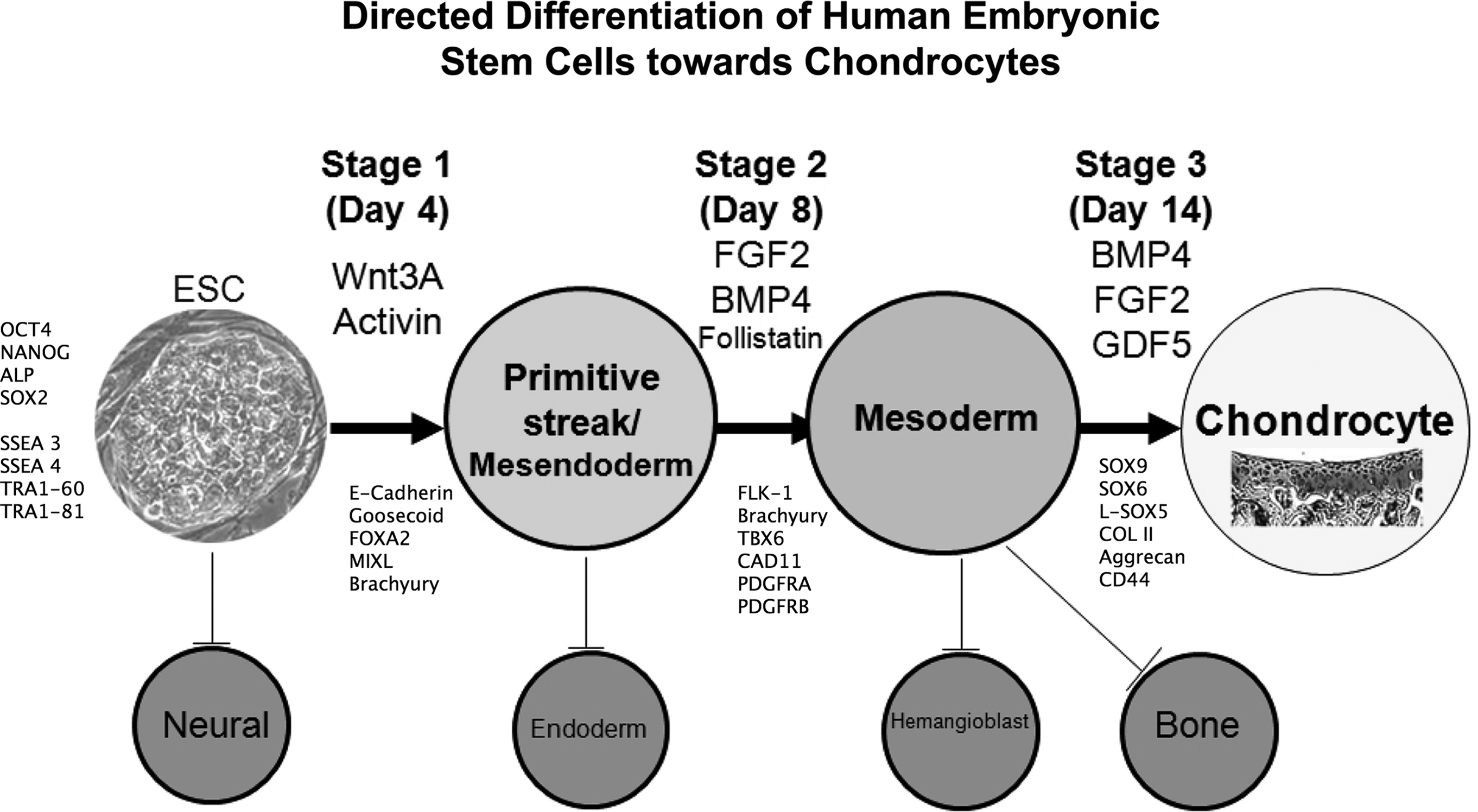
Schematic showing a breakdown of the defined protocol from our lab for directed differentiation of hESC to chondrogenic cells in three stages (modified from Oldershaw et al. 113 ).
ESC, embryonic stem cell; MSC, mesenchymal stem cell; GMP, good manufacturing practice.
Interestingly, a large-scale production of murine ESC-derived chondrocytes on microcarriers in serum-free medium has recently been reported. 117 This may be suitable for use in combination with our method to allow a rapid scale up the hESC-chondrocytes for clinical applications.
Tissue engineering using chondrocytes derived from pluripotent cells
Scaffolds or gel carriers play an important part in enhancing repair of cartilage by providing support for either endogenous cell-based repair or repair driven by implanted cells. Such scaffolds may also directly induce cell differentiation or maturation and facilitate cell survival in the damaged or diseased tissue. Obviously, rigid scaffolds such as those made from poly glycolytic acid, poly vinylalcohol, poly caprolactone (PCA), or poly lactic acid (PLA) provide more support under load, which may be particularly important after initial surgery, but may interfere with the properties of the maturing chondrocytes and hyaline cartilage if they do not show appropriate breakdown kinetics in the body and require more invasive surgical implantation. Poly(lactic-co-glycolic acid) and PLA scaffold has been used to support ESCs for differentiation to chondrocytes to make cartilage in vitro. 118 EB-derived hESCs seeded onto polycaprolactone scaffolds did not yield a high density of chondrogenic cells, 119 while hiPSCs from OA patient-derived synovial cells have been shown to form cartilage in a 3D PCA scaffold. 120 An alternative is to use natural or synthetic hydrogels in which the cells can be homogenously distributed before gel polymerisation and that are highly permeable though lacking in the strength under load of rigid scaffolds. While early studies embedding EB-derived chondrogenic cell in agarose showed poor viability, 121 a recent study showed that murine iPSC-derived and selected chondrogenic cells subjected to micromass culture and embedded in agarose were able to produce good hyaline cartilage-like tissue in vitro. 104 Encapsulation of EBs in a polyethylene glycol (PEG)-based hydrogel leads to cartilagenous tissue in vitro after TGF β1 treatment, 122 while derivitization of PEG diacrylate-hydrogels with peptides containing the integrin binding, arginine-glycine-aspartic acid fragment gave better cell adhesion for hESC-EB derived MSCs and those co-cultured with chondrogenic cells.123,124 The former cells embedded in a PEG diacrylate scaffold produced cartilagenous matrix in vitro and after subcutaneous implantation in nude mice. Cells within derivatized PEG diacrylate were significantly better than those in non-derivatized PEG diacrylate with regard to the quality of cartilage matrix produced. 93 Cartilage tissue engineering using chondrocytes derived from ESCs has also been achieved by implanting chondrocytes encapsulated in a HA-based hydrogel 86 into surgically induced cartilage defects in rats. This latter study showed characteristic remodeling, leading to good infilling of hyaline-like tissue containing collagen II and chondroitin sulfate-based ECM with negligible collagen I or X except in the subchondral layer (where extensive new bone formation was obvious) by 12 weeks. Importantly, repair was significantly better when hydrogels containing cells were used rather than gels alone. A chemically crosslinked dextran-poly(ethylene glycol) hydrogel has also been used for cartilage tissue engineering with an optimal degradation time. 125 hESC-MSC-seeded bilayer collagen scaffolds also showed the ability to generate cartilage in the patella groove defect area of rats. 126 Chondrogenesis from hESCs in perfusion bioreactors using porous silk scaffolds 127 and on nanoscale electrospun fibres has also been reported. 128 In general, none of these approaches has yielded flawless repair from hESC-derived chondrocytes and further research is needed, but the hydrogel studies, in particular, produce some quality hyaline, rather than fibrous cartilage in vivo, suggesting a way forward to clinical application.
Conclusions and Prospects
The temporal sequence of culture conditions has now been developed to induce hESCs to generate a relatively pure chondrogenic population without off-target differentiation, or any residual pluripotent hESCs. This is in a chemically defined format, suitable for larger-scale production for clinical applications. However, whether these cells can form functional hyaline articular cartilage with the necessary biomechanical properties, especially for long-term function, is still unknown. The immunological barriers are also an issue for ESC-based cell replacement therapy: novel regimes to prolong acceptance of ESC-derived tissues with minimal use of immunosuppressive drugs will speed up the translation of research to clinic. It is also as yet unclear what degree of tissue matching is needed for the use of allogeneic hESC derived chondroprogenitors in joint repair for human patients. Whether immunomodulation in parallel with hESc-derived chondroprogenitors will be advantageous to enhance the success of repair is another question that remains to be answered. By damping the endogenous inflammation at the osteoarthritic joint, we would envisage greater success in establishing repair cartilage in damaged joints. 129
Exploitation of human iPSCs will provide a further source of pluripotent cells that could be derived from patients' cells and raises the prospect of autologous treatment. Obviously, patient-specific iPSC-derived cell therapy should avoid immunorejection after transplantation. Thus, more research is needed on iPSCs technology so that it can be proved safe for therapy. However, the iPSC route to chondrocytes also opens the way to studying chondrocytes of defined genetic background and understanding how some genetic risks affect chondrocyte performance in different chondrogenic models in vitro. Recent studies showed that by overexpressing c-myc, klf4, and sox9, mouse dermal fibroblasts could be induced to give rise to chondrocytes that made hyaline cartilaginous tissue in vivo. 130 Although the cells produced so far showed karyotypic instability during culture, further optimization to overcome this problem may result in an alternative cell resource for cartilage engineering. Another concern for clinical application of ESCs-derived differentiated cells is retention of a small number of undifferentiated pluripotent cells that might give rise to teratomas. Interestingly, a compound, PluriSlns, which interferes with oleic acid biosynthesis, has been identified to selectively eliminated hPSCs while sparing a large range of progenitors and differentiated cells. Application of this compound during tissue repair would be predicted to increase the safety of PSC-based therapies. 131 It is very challenging to regenerate articular cartilage to a state that is functional and structurally able to withstand full joint movement and biomechanical loading. Expertise from biology, biomaterials, and the clinic are needed to devise ways to create precartilage tissue ex-vivo for implantation, or to deliver chondrogenic cells to form cartilage in vivo. The supply of chondrogenic cells is now robustly established, which opens up many possible routes for understanding cartilage defects, for drug development, and for regenerating cartilage using cell-based treatments.
Footnotes
Acknowledgments
The authors acknowledge funding for their work from the North West Science Fund UK, the NHS-BRC Manchester, and Arthritic Research UK and Chloe Duval for copy editing.
Disclosure Statement
No competing financial interests exist.