
Abstract
“Evaluating the Past and Present of Regenerative Medicine (RM)” was the first part of an Industry Symposium dedicated to the subject during the 2015 TERMIS World Congress in Boston. This working session presented a critical review of the current RM landscape in Europe and North America with possible projections for the future. Interestingly, the RM development cycle seems to obey the Gartner hype cycle, now at the enlightenment phase, after past exaggerated expectations and discouragements, as suggested by increasing numbers of clinical trials and recent market approvals of RM solutions in both Europe (Glybera and Holoclar® from Chiesi Pharma and Strimvelis® from GSK) and Japan (Remestemcel-L from Mesoblast®). The successful commercial translation of RM research is governed by five major drivers: (i) fully validated manufacturing capability for autologous or allogeneic products, (ii) reimbursement for targeted clinical indications with high and demonstrable medico-economic benefits versus standard of care, (iii) implication of regulatory bodies in the design and development plan of any RM solution, which should be well characterized, robust, with proven consistent efficacy and an acceptable and controlled positive benefit/risk ratio, (iv) collaborations facilitated by multicompetence hubs/consortia of excellence, (v) well-thought-out clinical development plans for reducing the risk of failure. Benefiting from past and present experience, the RM burgeoning industry is expected to accelerate the market release of cost-effective RM products with real curative potential for specific clinical indications with high unmet needs. This should be achieved by wisely leveraging all possible synergies of the different stakeholders, for example, patients, clinicians, reimbursement and health technology assessment (HTA) agencies, regulatory authorities, public/private investors, academia, and companies.
Introduction
E
A summary of the lectures and dialogues is presented here with an emphasis on five identified RM drivers: Manufacturing, Reimbursement policies, Regulatory environments, Collaborative development models, and Clinical evaluation platforms. In this review, the generic term, Regenerative Medicine, includes Advanced Therapy Medicinal Products (ATMPs), that is, medical products based on genes (gene therapy medicinal products), cells (somatic cell therapy medicinal products), and tissues (tissue-engineered medicinal products).
What We Have Learned from the Past
In the late 1970s and early 80s, Rheinwald and Green were able to grow serially passaged keratinocytes from a single cell suspension, 1 Bell et al. produced a tissue-like structure in vitro by contraction of collagen lattices using human fibroblasts 2 and, Yannas and Burke designed the first bilayer artificial skin composed of a temporary silastic epidermis and a porous collagen-chondroitin 6-sulfate fibrillar dermis. 3 These three major discoveries paved the foundation of the RM field and led to the commercial development of three products used in the treatment of wounds. Developing RM products is a long and difficult process. Like any other industrial products, a commercial cell-based therapy product must make financial as well as medical sense to be accessible to patients (Fig. 1).

We can learn from past experiences particularly when we look at the emergence of the therapeutic monoclonal antibody industry, which required ∼25 years to mature, but eventually produced the top-selling blockbusters of the pharmaceutical industry. 4 This coming of age of monoclonal antibody platform technologies can be delineated in terms of innovation S-curves 4 (Fig. 2). Such a retrospective analysis is particularly useful as it establishes a predictive model for the advances that still need to occur for RM to enable the generation of commercial products with superior efficacy and safety attributes. The humanization of chimeric antigen receptor (CAR) T cells —that is consisting in the modification of nonhuman protein to increase their similarity to human variants—constitutes an obvious learning from the process of therapeutic monoclonal antibodies.4–6

Emergence of the therapeutic monoclonal antibody industry (1975—horizon 2020).
From the experience of therapeutic monoclonal antibodies product development space exhibiting the following six dimensions can be forecasted for stem cell-based RM: (i) stem cell types (including allogeneic vs. autologous), (ii) manufacturing, (iii) efficacy enhancements, (iv) formulation, (v) transplantation and delivery, and (vi) stem cell therapeutics combined with conventional pharmaceuticals. 4
RM Current Status
The market of RM, estimated by industry analysts, is estimated to reach US$24.7 billion by 2017. 7 This figure includes not only revenues from RM (i.e., ATMPs as defined previously in the “Introduction” section), but also revenues from cytokine and growth factor therapies as well as tissue grafts with regenerative properties. The orthopedic indications market alone has been projected to reach US$10.3 billion by 2017. Currently, North America dominates the global market due to heavy investment in the development of regenerative products as well as higher numbers of commercialized products. North America represents more than 50% of the regenerative companies worldwide compared with 27% for Europe and Israel. 8 However, research and development in Europe, Japan, and South Korea is now picking up pace.
Continued development of the RM field is expected to be driven by the efficacy of currently proposed solutions including: (i) registered products like Carticel® (Vericel Corporation) in the United States, which consist of autologous cultured chondrocyte therapies indicated for the repair of symptomatic cartilage defects of the femoral condyle (medial, lateral, or trochlea), caused by acute or repetitive trauma, (ii) products in development in the United States such as TEMCELL®, formerly known as Prochymal® (Remestemcel-L; Mesoblast®), a cell therapy product for the treatment of graft-versus-host disease (GvHD), a major complication of bone marrow transplants. TEMCELL, with regulatory approval in Canada, New Zealand, and Japan targets a small population of patients, but shows a very high-level response rate, +90%, with substantial increase of life expectancy. 9
The RM industry is now more seriously entering its commercialization stage. Therapeutic living cell products have been developed in three waves: (i) vaccines, blood transfusions, or hematopoietic stem cell transplantations, (ii) epithelial, bioengineered skin, and chondrocyte-based autologous and allogeneic therapies, (iii) genetically modified T cells for cancer therapy or other diseases, now fast approaching the market with clinical trials rapidly increasing, both in number and in size (i.e., average number of enrolled patients per trial) (Fig. 3).

Exponential development of genetically modified T cells in cancer therapy.
Three major clinical milestones were achieved in 2015: (i) CAR T cell product candidates JCAR014 and JCAR015 developed by Juno Therapeutics demonstrated encouraging clinical responses in patients with relapsed or refractory lymphoblastic leukemia, (ii) the CAR T cell product CTL09 developed by Novartis in collaboration with the University of Pennsylvania demonstrated, in a Phase II clinical study, a stunning 93% complete remission in pediatric patients with relapsed or refractory acute lymphoblastic leukemia, and (iii) gene-edited immune cells UCART19, a product comprised of allogeneic T cells engineered using the TALEN gene editing tools developed by Cellectis, have been used to successfully treat an otherwise incurable acute lymphoblastic leukemia in a 1-year-old child.10–13
Several other RM products currently in Phase III clinical trials will read-out in the next 2 years, including, in 2016 alone, the following (not exhaustive list): (i) CHART-1, an autologous stem cell preparation developed by Celyad for congestive heart failure, (ii) MSC-100-IV, an allogeneic mesenchymal lineage stem cell preparation developed by Mesoblast for acute pediatric GvHD (a similar product, TEMCELL, was fully approved at the end of 2015 for adult and pediatric GvHD patients in Japan), 14 (iii) MPC-150-IM, a mesenchymal precursor cell preparation developed by Mesoblast in partnership with Teva for chronic heart failure, (iv) MPC-06-ID, another mesenchymal precursor cell preparation developed by Mesoblast for chronic low back pain due to degenerative disc disease,15,16 and (v) Tigenix Cx601 for the Treatment of Complex Perianal Fistulas in Patients with Crohn's Disease, currently under evaluation by Emergency Medicines Agency (EMA). In addition, the Phase III gene therapy clinical trial of SPK-RPE65 from Spark Therapeutics, developed as a treatment for RPE65-mediated inherited retinal dystrophies, is also expected to read-out at the end of 2016. 17
Based on the current batch of ongoing clinical trials and including an estimated success rate for each of the different phases of translational development, the market for stem cell-based therapies alone has been estimated to grow at a compound annual growth rate (CAGR) of 70% from US$5 million in 2012 to nearly US$9 billion by 2025. 18 Furthermore, given the stunning efficacy in cancer already observed using the first generation of CAR T cell products, one can expect that the RM market as a whole, including CAR T cell therapeutics, will be a multiple of the abovementioned forecast stem cell-based therapy market (Fig. 3).
Importantly, such advances need to be backed up by the creation of tangible and monetizable intellectual property (e.g., patents, manufacturing secrecies) to create a sustainable and trustworthy innovation ecosystem, where companies and investors have a suitable horizon for generating appropriately rewarding payoffs.
The Five Drivers of RM
Manufacturing
There is a growing need for strong cell therapy manufacturing capacity and, in turn, anticipation of the future total global manufacturing demand to avoid production capacity insufficiencies. Hopefully, lessons have been learned from the initial stages of commercialization of monoclonal antibodies and other therapeutic proteins, where manufacturing capacity shortages were experienced during the early years, thereby creating a serious drag on the biotech industry, since the building of new manufacturing plants typically take 3–5 years. 19
Novartis secured access to a dedicated and validated FDA-approved cell manufacturing plant by acquiring in 2013 the $43 million New Jersey-based plant that Dendreon had been using for manufacturing its autologous treatment Provenge (Provenge was sold to Valeant Pharmaceuticals International for $415 million). 20 This facility was used to produce the clinical trial lots and cover the anticipated commercial demands for Novartis's first autologous CAR T cell treatment. In addition, Novartis created a dedicated cell and gene therapy unit in 2014, and that same year, invested €15 million ($20 million; average currency rate in 2014: 1 € = 1.33 US$) for building a 30,000-sq.ft. facility for T cell R&D and manufacturing at the Penn Medicine medical center.21–25
Access to a commercial-scale manufacturing plant, with the aim to produce not only GMP-compliant clinical trial lots, but also commercial treatment doses, has likewise been a strategic focus of several of the leading biopharmaceutical companies developing cell-based RM solutions. This includes Geron who constructed a manufacturing suite on its industrial site for producing regenerative therapies derived from pluripotent stem cells, as well as Athersys and Pluristem to produce therapeutic products based on cells of the mesenchymal lineage. 26
Moreover, in 2011, Mesoblast entered into a strategic alliance with the Lonza Group, a leading biologics contract manufacturing organization, to produce allogeneic adult mesenchymal precursor stem cells. The agreement provides Mesoblast with the certainty of capacity to meet its long-term commercial product needs through a process whereby Mesoblast can trigger Lonza to construct a purpose-built manufacturing facility exclusively for producing Mesoblast's marketed products. Mesoblast will have the option to acquire this plant at a pre-agreed price 2 years after the facility receives its approval by the regulatory authorities. 27
Another remarkable indicator that manufacturing capacity has become a key strategic capability is further demonstrated by WuXi Apptec, a worldwide contract research and manufacturing organization, which has started to offer R&D and manufacturing services for the emerging RM industry. They have constructed, in response to increasing customers' demands, a 45,000 sq.ft. facility in Philadelphia with advanced modular design suites, complementing an existing 16,000 sq.ft. for cGMP cell therapy manufacturing, to produce both autologous and allogeneic cytotherapies. 28
Reimbursement
Each development stage of a RM product, including the earliest ones, must be well-thought out in terms of the data that should be generated to support the product value. This is especially true considering that the headroom may change during the product development timeline with new competitor entrants, changes in the standard of care, and pressure on healthcare expenses and costs. Target product profiles and early discussions with reimbursement bodies to understand their expectations need to be considered as manufacturing costs, and reimbursement issues may remain high for cell-based therapies. This was illustrated by the commercial failure of Dendreon shortly after the launch of Provenge (sipuleucel-T).20,29 Because CAR T cell therapies are expected to be one-time treatments, it is anticipated that they will be quite expensive and may cost $450,000 or more.23,24 More clinical data and experience with such treatments are still required to anticipate the reimbursement level, especially considering the fact that some of the patients treated with CTL019 had experienced severe complications such as cytokine release syndromes that can be lethal if left untreated.22,23 Anaphylaxis represents another type of adverse event observed with CAR T cell therapies that will be ultimately prevented by further technological advancements such as the use of fully humanized CAR antibody constructs.5,6 CAR T cell technology is indeed evolving very quickly leading to a new generation of safer products. Possible advances currently being explored include the use of bifunctional small molecules as molecular “switches” to redirect and regulate genetically engineered CAR T cells, and the use of a T cell construct with an inducible suicide gene relying, for example, on rimiducid-inducible caspase 9 activation.30,31
As for cytotherapies exemplified by Holoclar®, an autologous cell therapy for the treatment of thermal or chemical burns to the eye and the first stem cell-derived medicinal product was approved in Europe. Another example is TEMCELL HS Inj. for the treatment of acute GvHD, which is the first stem cell therapy approved in Japan and priced at up to ¥20 million per course of treatment (∼US$170,000), it is expected that the price of gene therapies will be very high. Glybera, a potentially curative treatment for lipoprotein lipase deficiency and the first gene therapy medicinal product approved in Europe, has been priced in Germany for its first year of commercialization at €1.1 million per average course of treatment. The product is, at this price, the most expensive treatment for a rare disease32–34 to date. Such high prices remain controversial and will obviously not be sustainable for national healthcare systems when it comes to diseases of higher incidences.35–37
Because of their high manufacturing costs and prices, as well as their infancy stage and thus their still uncertain market penetration rates, cell therapies need to demonstrate clinical benefits that no conventional product can deliver. This fact was clearly demonstrated in the case of Provenge that quickly met with strong competition from the oral medicines Zytiga commercialized by Johnson & Johnson, and from Xtandi marketed by Astellas and Medivation 20 soon after its launch of a hormone-resistant prostate cancer treatment.
Regulatory
The regulatory process, in so much as it plays a critical role in facilitating innovation and early access to the market, is another important element impacting the development of the RM industry. A simpler process based on an accelerated, conditional approval for cell-based medicine, as enacted in 2014 in Japan, notably creates incentivizing conditions for biopharmaceutical companies to invest and seek product approval for new medicines. 38 According to a press release by Chiesi about its spin-off Holostem, and particularly about the 2015 regulatory approval for Holoclar in Europe, the industry and regulatory authorities are still in a learning mode when it comes to human cell-based medicinal products: “The authorization process has been long and complex, but the result achieved today shows that cells can be cultured according to pharmaceutical standards appropriate to guarantee safety and efficacy ……. In addition, in a period of great confusion about the real therapeutic possibilities of stem cells, such as the one we are living in, being able to demonstrate that stem cells can be definitely safe and successful in a controlled clinical setting is more important than ever”. 39
To this date, there are only seven RM products (ATMPs) approved in Europe: ChondroCelect (TiGenix; marketing authorization recently withdrawn for commercial reasons, in July 2016), Glybera® (uniQure/Chiesi), MACI (Vericel Corporation; market approval suspended after the closure of the EU manufacturing site for commercial reasons), Provenge (Sipuleucel-T; Dendreon) (marketing authorization withdrawn at Dendreon's request), Holoclar (Holostem/Chiesi), Strimvelis (GSK), and Imlygic® (talimogene laherparepvec; Amgen) (Fig. 4). In addition, there are several other RM products delivered to patients under the Hospital Exemption scheme.
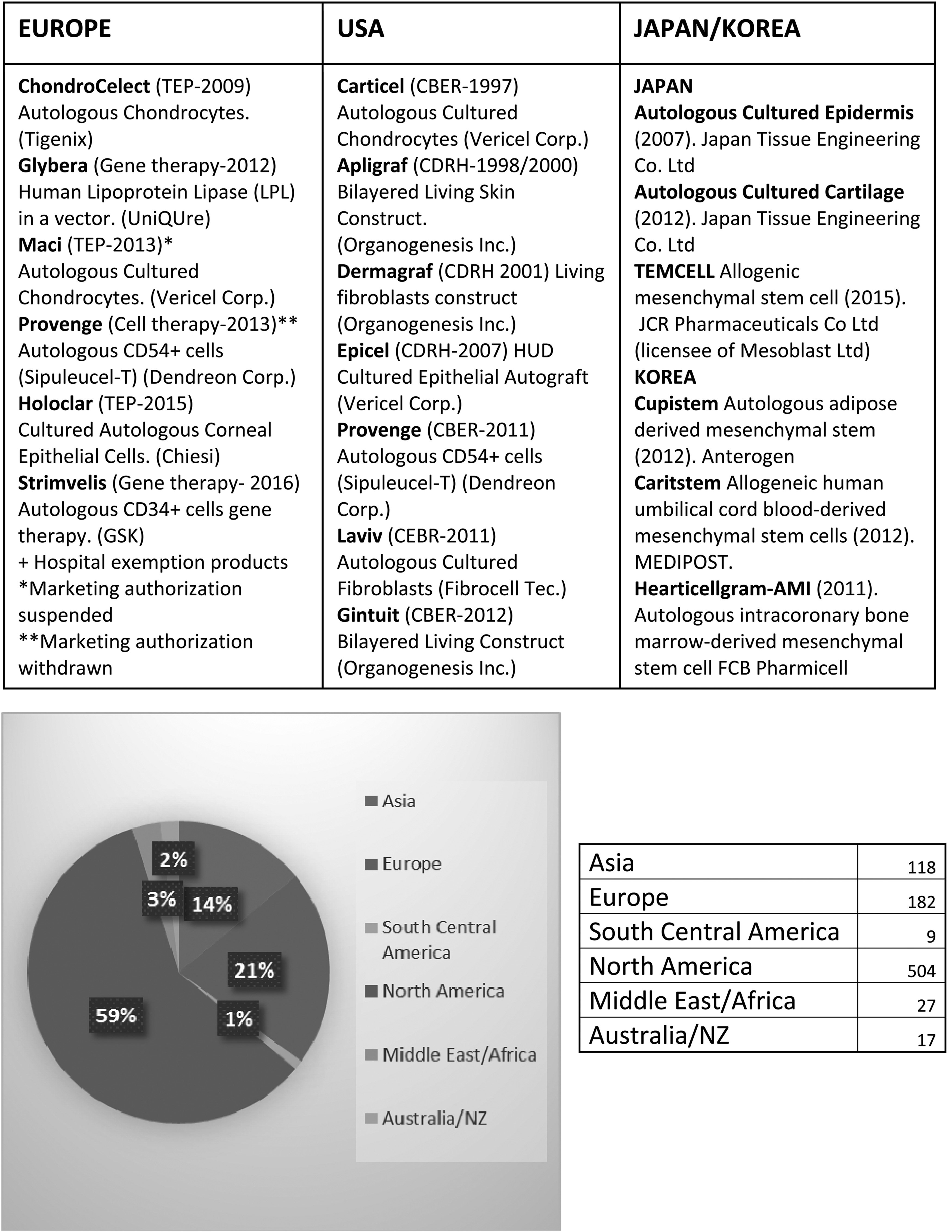
Current status of cellular therapies.
Two legal instruments dominate the regulation of RM in the EU: the European Tissue and Cell Directive (EUTCD) and the ATMP regulation. EUTCD aims at setting standards of quality and safety for the donation, procurement, testing, processing, preservation, storage, and distribution of human tissues and cells. It is applicable for human cells and tissues used for therapeutic purposes, in case of homologous use and minimal manipulation. It does apply as well for the starting cell and tissue materials of ATMPs. ATMP regulations aim at regulating tissue-, cell- and gene-based therapy medicinal products (in such case, cells and tissues are either substantially manipulated or intended for nonhomologous use). Another important consideration is that EU law applies at two levels: regulations are applied directly, whereas directives are applied through the implementation of legislation in each Member State. 40
For example, the EUTCD directive needs to be implemented through national laws. This can lead to some discrepancies between Member States. On the contrary, ATMP regulation applied as such governs those products needing mandatory centralized approval through the European Medicine Agency. 40 There are notable exemptions to the requirement for a mandatory centralized marketing authorization. In particular, “the hospital exemption” allows for a product to be authorized at the national level if it is (i) prepared on a nonroutine basis according to specific quality standards, (ii) used within the same Member State as that in which it is prepared, and (iii) used under the supervision of a medical practitioner pursuant to an individual medical prescription to the patient. 40
Nonetheless, the European Commission published an important report in 2014 on European Directive 2001/83/EC and regulation 726/2004, amended by regulation 1394/2007 that sets the regulatory framework for ATMPs and further clarifies the recommended use of hospital exemption. In this report, the European Commission clarifies through recommendations and conclusions that aim to preserve R&D investment incentives while permitting hospital exemptions, notably for product use in individual patients. 41
Possible regulatory shortcuts in Europe include the streamlined product development plan based on the Risk-Based Approach (Annex I, Part IV of Medicinal Product Directive 2001/83/EC), taking into account “the identification of various risks associated with the clinical use of an ATMP and risk factors inherent to the ATMP with respect to quality, safety, and efficacy” and which acknowledges that risks are specific to each particular product and subject to an array of factors, including the biological characteristics of the product, the manufacturing process used to produce it, and the specific therapeutic use(s) of the ATMP.
Another pilot initiative for rapid access to the market is the Adaptive Licensing pathway (EMA/254350/2012), where the initial authorization is based on an early risk/benefit assessment in a small population (to be extended in an iterative process) or based on surrogate primary endpoints (conditional approval). This initiative reflects the fact that regulators have increasingly moved toward a proactive attitude beyond simply evaluating products.42–45 The EMA defines adaptive licensing as follows: “a prospectively planned, adaptive approach to bringing drugs to market. Starting from an authorized indication (most likely a “niche” indication) for a given drug, through iterative phases of evidence gathering and progressive licensing adaptations concerning both the authorized indication and the potential further therapeutic uses of the drug concerned Adaptive Licensing seeks to maximize the positive impact of new drugs on public health by balancing timely access for patients with the need to provide adequate evolving information on benefits and harms”. 46
The regulatory environment in the United States is based on the classification of the RM products as biological product, drug, or device, for respective assignment to one of the three FDA centers, the Center for Biologics Evaluation and Research (CBER), the Center for Drug, Evaluation, and Research, or the Center for Devices and Radiological Health (CDRH). The assigned Center retains the primary regulatory responsibilities for product filing review and approval. The examination is driven by a risk-based approach and enabled approval of RM products earlier than in Europe, with Apligraf, a tissue-engineered skin and Carticel, autologous cultured chondrocytes, approved as early as 1997/1998, respectively by CDRH and CBER (see Fig. 4).
The riskiest RM categories are: gene therapy, nonautologous and manipulated cells and tissues. The market approval can be more challenging for combination products, defined as products comprising two or more regulated products such as tissue-engineered products. This has been illustrated by matrix-induced autologous chondrocyte implant (MACI), developed initially by Genzyme, and associating cultured cells and biomaterials. MACI has not yet been approved by the FDA for clinical use, although it was approved in EU back in 2013. Vericel Corporation, who acquired the product from Genzyme, has just recently received FDA acceptance, in March 2016, for Biologic License Agreement filing 47 after multiple interactions with the FDA—the product is currently under evaluation. It is interesting to note that MACI was first submitted for market authorization in Europe, in September 2011, with approval in March 2013. Since then, the market approval has been suspended after the closure of the EU manufacturing site for commercial reasons, but not for safety concerns. 48
It is strongly recommended to ask for frequent consultations with the Regulatory Authorities throughout the development, from before first-in-human study to the end of Phase II, to build a solid registration study. One big advantage of the United States system compared with that of Europe is that in the United States, discussions are made with a single agency compared with multiple regulatory bodies in Europe. Product development being very often global, the EMA and FDA have fortunately regular meetings, every 2 months, for discussions on most challenging issues related to cell, tissue, and gene therapy medicinal products to try to harmonize their approaches. Now that Japan has extensively revised their RM product guidelines, Japan may now become a first target for companies considering possibility of early market access with conditional approval (Fig. 4).
To conclude, four key recommendations that could expedite the regulatory assessment of a RM product: (i) develop a cluster of RM products with possibilities of expediting/fast tracking market approval approaches, (ii) make the product as simple as possible to facilitate tracking and consistency, (iii) put in place robust quality assurance/quality control (QA/QC) controls before moving to clinical trials including potency assays to assess functionality, support comparability between formulations and justify evolution between products and the definition of precise biomarkers to characterize the product and surrogate endpoints to predict clinical efficacy, (iv) justify in-depth the risk-based approach.
Collaborative models
Hubs of excellence
The emergence of RM and gene therapy as an economic sector has further materialized in the creation of hubs of excellence, where scientific competences in various academic disciplines converge with a variety of economic actors, including venture capital firms, consulting firms, and contract research and manufacturing organizations. Examples of such hubs in Europe are the Cell and Gene Therapy Catapult of London, the London–Cambridge–Oxford golden triangle, the Scottish stem cell hub of Edinburgh, CABIMER in Spain, and clusters in Leipzig and Berlin-Brandenburg. Remarkably, pan-national consortia have also been created to benefit from regional synergies exemplified by the Bioreg project that regroups three universities, three business clusters, a start-up center, and a foundation, thereby representing Spain, Portugal, and the south of France.
Furthermore, collaboration within various European countries can take many forms, as exemplified by Holostem Terapie Avanzate, a partnership spin-off founded in 2008 by Chiesi Farmaceutici s.p.a. and the University of Modena and Reggio Emilia. At present, Chiesi is developing both alipogene tiparvovec (Glybera), the first gene therapy approved in Europe, and GPLSCD-01 (Holoclar), the first European-approved stem cell therapy. Glybera was developed by the gene therapy biopharmaceutical company uniQure and licensed to Chiesi for €17 million in fees and €14 million in equity financing for its commercial rights in Europe and to codevelop uniQure's gene therapy for hemophilia B. 49 Holoclar was developed by Holostem. This remarkable success takes its roots not only in the engagement of the region to support innovation, but also in synergistic innovation-promoting corporate and academic cultures. 50 Chiesi expresses this culture through concepts of “value” and “innovation entrepreneurship” as follows 51 :
Value
“Our drive is to generate value today and in the future through entrepreneurship and innovation. Entrepreneurship means having the competencies to take the responsibility and calculate the risk to challenge the present and grasp opportunities that are in line with our principles. Innovation means the capability to continuously change through a consistent process of appreciating the value of intuition and the generation of ideas coming from within our professional community”
Innovation entrepreneurship
“Express energy, proactivity and enthusiasm to compete with the best-in-class and win the challenges that come up every day. Encourage and reward actions and the development of new ideas, accounting on competencies, intuitions, and opportunities. Promote decision and risk taking, as well as the courage, intended as the engine to generate change and continuous improvement.”
Example: Center for Commercialization of Regenerative Medicine
The road to development of new technologies and products is paved by a number of hurdles, including valleys-of-death in the quest for funding, complex patent and freedom-to-operate landscapes, and the identification of optimal partnerships and business models. In parallel, companies are generally created too prematurely with the lack of appropriate resources.
Center for Commercialization of Regenerative Medicine (CCRM), created following this assessment, aims to help companies through accelerated funding of the early stages development. Time-to-market can no longer take 25 years as was previously the case. The CCRM was founded through a unique Canadian government program (the networks of Centers of Excellence) to build the right kind of ecosystems and networks to drive technologies and products to commercialization. CCRM works with a strong academic network of stem cell and RM scientists in Canada. The industry consortium is composed of more than 40 companies worldwide. CCRM aims to build a global network, in a nonprofit set-up, and to drive technologies forward.
In June 2015, CCRM launched its first company, ExCellThera, offering cell therapy assets for the treatment of acute leukemias and other blood cell disorders. CCRM and ExCellThera have developed an innovative method for faster expansion of hematopoietic stem cells from cord blood, which should offer advantages over bone marrow transplants in terms of infection and immunological complications and cost effectiveness. The Phase I/IIa clinical trial began at the end of 2015. In addition to incubating a number of other companies not yet launched, CCRM has invested in a manufacturing facility, not simply aiming to produce cells, but do so using innovations in closed systems, automation, and novel upstream and downstream processes. 52
After 3 years of operations, CCRM is now trying to build the investor network by attracting companies to the ecosystem with unique assets such as: (i) internal manufacturing capabilities, (ii) dense and very active clinical centers in Toronto and neighborhoods, (iii) facilitated access to clinical centers for clinical evaluation, (iv) a positive Canadian regulatory environment with a history of 1st worldwide approvals (e.g., Apligraf, TEMCELL), and conditional/fast track regulatory approvals in Canada.
The CCRM coordination of translation and commercialization is already of benefit for the academic consortium. CCRM has been chosen as the commercialization partner for RM technologies developed under “Medicine by Design,” a +100 million Canadian Dollars federal investment.
CCRM V2.0
CCRM is also invested in promoting local hubs around the world for translation and commercialization of RM solutions by academia and industry consortia. This has been recently illustrated by the creation of Regenerative Medicine Coalition with leading core members in RM translation (i.e., Wake Forest | Institute for Regenerative Medicine; CABIMER | Andalusian Center for Molecular Biology and Regenerative Medicine, Sevilla, Spain; Advanced Center for Biochemical Engineering, University College London, London, United Kingdom; CCRM | Center for Commercialization of Regenerative Medicine, Toronto, Canada; MIRA | Institute for Biomedical Technology and Technical Medicine, University of Twente, Enschede, Netherlands; McGowan Institute for Regenerative Medicine, Pittsburgh, PA; and BCRT | Berlin-Brandenburg Center for Regenerative Therapies, Germany). Strategic alliances have also been contracted by CCRM with Cooperative Research Center for Cell Therapy Manufacturing, Australia and Cell Therapy, Catapult, United Kingdom.
The global network of local hubs is expected to enable/enact the development and commercialization of cell therapies on a global level, the fast track development of cell therapies for a global market.
Example: the EU framework program for research and innovation, horizons 2020
The European strategy for healthcare, and its associated Framework Program 8, constitutes powerful instruments that build upon the achievements of the previous European science & technology strategic plans and Framework Programs 6 and 7. At present, about 30% of RM and gene therapy companies are located in Europe. A finer analysis further reveals that most of the stem cell-based projects conducted under Framework Program 6 have been performed in Germany and in the United Kingdom. Notably, the EU has financed, under Framework Program 7, not only clinical projects, but also preclinical projects targeting major unmet clinical needs. 50 For example, the program REGENER-AR received EU-sourced funding of 5.9 million Euros for clinical trials using stem cells for rheumatoid arthritis treatment.
Preclinical development projects have included: THYMISTEM (production of thymus cells for therapy; 6.0 million Euro funding); PLURIMES (production of bone and muscle-forming cells; 6.0 million Euro funding); and, NEUROSTEMCELLREPAIR (cell replacement therapies for neurological disorders, e.g., Parkinson's disease; 6.0 million Euro funding). Consequently, leading European nations with a strong biotechnology sector such as the United Kingdom or Germany have been actively seeking to generate competitive advantages by investing heavily in this emerging sector. 50
These efforts are starting to provide visible payoffs. An example of company creation is Orbsen Therapeutics Ltd., which was founded in 2006 as a spin-off from the Ireland's Regenerative Medicine Institute. Notably, Orbsen participated under Framework Program 6 in the DECIDE project in the field of hematopoiesis and cancer, and in the REDDSTAR project under Framework Program 7 to study microvascular complications of diabetes. What is more, its core technology consisting of selecting CD362- (syndecan-2) bright plastic adherent cells as a more homogeneous therapeutic preparation of cells of the mesenchymal lineage, was derived from the PURSTEM project funded through Framework Program 7. 53 The high level of productivity of European researchers provides a solid foundation to achieve many more similar successes.54–55 However, the EU has fewer young firms among its leading innovators as compared with the United States; this accounts for about a third of the corporate R&D intensity differential between these two macroeconomic regions and reveals a structural issue. 56
Given that the coming of age of RM, gene therapies, and cytotherapies bear, beyond clinical benefits for patients who need them, important economic benefits, macroeconomic perspectives of emerging segments of the pharmaceutical industry are becoming evident. As such, RM represents an engine of economic transformation of the 2010–2020 decade. 38 From the point of view of the pharmaceutical and biotechnology industry of the EU, which has long lagged behind that of the United States in terms of investment and economic returns, this emerging segment of the healthcare industry constitutes an opportunity for European countries to build a competitive advantage by implementing a virtuous cycle of investments, job creation, and total factor productivity enhancements. The latter have the potential to generate superior profits for European companies in the field, increasing the global attractiveness of the region, and in turn triggering more investments. 38
Clinical trials
Phase III clinical trials, designed to confirm the efficacy results of Phase II trials in larger groups of patients, are the ultimate step whereby the safety, efficacy, and dosing of a given therapy is evaluated in humans. It is the point where research, manufacturing, and operations are pressure tested before going to large batch production and commercialization. These clinical trials are large, long, and costly. As they carry a lot of weight for companies and investors, it is important to manage them carefully. For example, for drug development, more than half of late Phase III trials fail to show sufficient efficacy or safety. 57
Several small molecules and cell therapy products have failed in phase III clinical trials after showing very promising results in phases I and II. In 2008, the Renovo Group announced positive results of Phase II clinical trials for two products based on human recombinant TGF-beta-3 for reduction of scarring. A few years later, the outcome of the phase III trials was defined as a “clear miss.” The trials had involved more than 350 patients who were given different doses of the drug at 56 centers in the United Kingdom, France, Hungary, Germany, Italy, Poland, Spain, Denmark, Latvia, and United States. 58
The same story occurred more recently with HP-802-247, a spray-on skin treatment consisting of living human cells made by Smith and Nephew, designed to help heal leg ulcers. This product failed in clinical phase III, which was unexpected given the promise of earlier phases I and II results. 59 What is the disconnect between promising phase I and II and failed phase III trials?
According to the European Center for Pharmaceutical Medicine, clinical trial failures can be summarized into six main categories: basic sciences, clinical study design, dose selection, data collection and analysis, operational execution, and others causes. 60 In addition to these different aspects, pressure to succeed may induce manufacturers and business developers to rush to Phase III once an exciting signal shows up at the Phase II stage. All of these problems can result in efficacy failures, safety failures, as well as commercial/financial failures.
Herreros et al. 61 published a posttrial analysis of their phase III failure for Fistula Advanced Therapy Trial 1, a study sponsored by Cellerix, aiming at evaluating the use of autologous adipose-derived stem cells for the treatment of complex fistula-in-ano. They identified possible causes of efficacy failures due to differences in inclusion criteria (e.g., severity of anal fistula), surgical procedures, cell handling, and cell formulation with or without fibrin glue. Interestingly, the Center with the most experience in dealing with fistula and cell therapies did actually show efficacy with the treatment. 61 Similarly, postmortem analyses should be shared and published to help improve the rate of success of phase III clinical trials.
The RM industry should learn from the experience of pharmaceutical companies for the prevention of clinical failures. Different approaches aiming at strengthening the quality control and the robustness of clinical trials in a generic way have been tried. They may be certainly applicable to the clinical evaluation of RM products while considering their specificities. AstraZeneca's “5R” approach to ensure Phase III work is designed to confirm “Right Target, Right Tissue, Right Safety, Right Patients, and Right Commercial Potential.” 62 Pfizer has put in place its Three Pillar framework 63 and Roche has further invested in computer simulations. 64
In addition, previous experience has shown the imminent importance of: (i) developing rigorous risk assessments and mitigation from the very beginning and during the overall development process (ii) designing Phase II trials to gather a maximum amount of information related to the product, the patient population and mechanisms of action (iii) setting up a well-organized project team with a strong leadership able to resist to pressure and able to communicate openly and frequently with all players from research to business (iv) using Target Product Profiles as a main driver during the full development path (v) optimizing Phase III study design, that is, protocol review and optimization, adaptive trial designs, and biomarkers (vi) using a multidisciplinary team to thoroughly analyze the protocol and the clinical trial execution involving internal and external expertise from regulatory, R&D, operations, and the commercial sector (vii) de-risking Phase III execution by carefully analyzing study execution risks and conducting ongoing surveillance of the quality of data being collected. 65 In addition, special attention should be paid on the characterization of the RM product to be evaluated and its quality control tests, to limit the risk of biases and failures, due to the product itself.
Concluding Remarks
In sum, accelerating the commercialization of RM products is an important aim, the realization of which will provide wide-ranging benefits. Cellular and gene therapies bring to patients and to clinical practitioners not only novel disease-modifying therapeutics, but also therapies that have the potential to be curative. Moreover, even if highly but appropriately priced (based on their societal values), these products will ultimately result in economic benefits for health systems by reducing the overall cost of managing certain diseases and particularly chronic diseases. 66 Patients will be enabled to live longer, healthier, and more productive lives, and thus, further contribute, including economically, to society and their professions. In addition, the rise of the new industry will enhance the economic capital of the jurisdictions where they are deployed, and particularly where they are deployed first all the way from R&D to manufacturing. Several regions have already met with tangible success in the emerging field of RM and gene therapy.
A further long-lasting, competitive advantage could be created by increasing even more public and private investments dedicated to such emerging fields of the pharmaceutical industry, by incentivizing further corporate R&D intensity through identifying and addressing structural impediments, and by increasing regional and/or worldwide coordination and collaboration, not only between companies or academia, but also between relevant regional authorities.
Footnotes
Acknowledgment
The authors are very grateful to Dr. M. Rathman for her advice and careful proof reading of the article.
Disclosure Statement
No competing financial interests exist.