
Abstract
Background/Aims:
The ability of endothelial progenitor cells (EPCs) to home to sites of neoangiogenesis makes them attractive candidates for use in the field of gene therapy. The efficacy of this approach depends on the efficiency of the vector used for transgene delivery.
Methods/Results:
In this study, we have compared the efficiency of adenovirus, five serotypes of AAV2, VSVG-pseudotyped lentivirus, and nonviral plasmid/liposome DNA vectors to deliver the green fluorescence protein reporter gene to human early EPCs to determine efficacy and vector-related cell toxicity. Adenovirus proved most effective with efficiencies of up to 80% with low levels of cell death. Lower levels of expression were seen with other vectors. Electroporation proved unsuitable at the parameters tested. We have also identified at least two distinct subpopulations that exist in the heterogeneous parent EPC culture, one of which is amenable to transduction with adenovirus and one that is not. In addition, adenoviral transduction did not disrupt the ability of the cells to incorporate into endothelial structures in vitro.
Conclusion:
We have found adenovirus to be the most efficient of the vector systems tested for gene delivery to EPCs, an effect that is mediated almost entirely by one of two identified subpopulations.
Introduction
The homing ability of these cells makes them attractive candidates for use in gene therapy after they have been genetically modified to specifically target areas of disease and carry potentially therapeutic genes, including to tumor cells that have a high rate of neoangiogenesis. 10 Their engraftment into sites of vasculogenesis could provide a more sustained level of gene expression over a longer period of time. To date, various studies have been carried out in which EPCs have been genetically modified using plasmid DNA, 11 adenovirus, 12 and lentivirus, 13 but to our knowledge there has been no direct comparison of vector efficiency or resulting vector-related cytotoxicity. In addition, no consensus yet exists as to the definition of an EPC, and many studies use different culture conditions, which further complicates attempts at direct comparison.
In this study, we have used both viral and nonviral vectors to deliver a reporter gene to human early EPCs. These cells have comparatively low proliferation rates, meaning cell numbers isolated from each donor or available for autologous transplant may be limited, a fact that potentially makes gene transfer to these cells all the more relevant. Indeed, it has already been shown that gene transfer has substantially lessened the number of cells required in vivo to produce comparable therapeutic effect. 14 Moreover, it has been shown that EPCs may be dysfunctional in disease states such as diabetes mellitus, 15 and, here too, gene transfer is a potential method to attenuate dysfunction in these cells for purposes of autologous transplantation.
Thus, we present a comparison of different viral and nonviral vectors for gene delivery to human early EPCs with consideration of the effect of the procedures on cell survival and subsequent functionality.
Materials and Methods
Patient characteristics
All procedures were carried out with approval from the Ethics Committee of University College Hospital, Galway. Peripheral blood was obtained from patients aged between 25 and 65 years undergoing venosection as part of treatment for hemochromatosis after informed consent was given. Previously published studies have used similar patient cohorts as normal controls in analysis of EPCs isolated from peripheral blood. 16 Only nondiabetic patients who were not taking medications and who were in a maintenance treatment program to ensure uniform low levels of iron in their blood were included in the study. These patients were otherwise healthy and had ferritin levels of less than 50 ng/mL (normal range 10–200) and were not anemic. Three donors were tested with each vector, and transductions/transfections were performed in triplicate for each donor.
EPC isolation and culture
Cells were isolated using a modification of previously described techniques.2,4,17,18 Whole blood was diluted in a 1:1 ratio with HBSS (Sigma, Arklow, Ireland) containing 1 mM ethylenediaminetetraacetic acid and 5% bovine serum albumin, and peripheral blood mononuclear cells (MNCs) were isolated by density gradient centrifugation by gently layering diluted blood over Ficoll Paque Plus (Amersham, Buckinghamshire, United Kingdom). MNCs were then washed in phosphate-buffered saline and plated on 12-well plates coated with human fibronectin (Sigma) at a density of 1 × 107cells/well. The cells were maintained in endothelial growth medium-2 (Cambrex, Slough, United Kingdom) supplemented with 2% fetal bovine serum, human epidermal growth factor, hydrocortisone, gentamicin, amphotericin-B, vascular endothelial growth factor, human fibroblast growth factor-B, R3-insulin-like growth factor-1, ascorbic acid, and heparin, and incubated at a temperature of 37°C and 5% CO2. Medium was changed at day 2 and daily thereafter. After 7 days, cells were harvested using standard trypsinization procedures. Endothelial phenotype was determined by uptake of acetylated low density lipoprotein (LDL) (Molecular Probes, Dublin, Ireland) and binding of ulex europaeus agglutinin-1 (UEA-1) lectin (Sigma, Arklow, Ireland), as previously published19–23 and expression of endothelial markers kinase domain receptor (KDR) and von Willebrand factor. Staining and analysis were carried out as previously described. 2
Adenovirus
Ad enhanced green fluorescence protein (EGFP) is a replication-deficient, E1- and E3-deleted adenovirus vector that expresses the EGFP gene under transcriptional control of the cytomegalovirus (CMV) immediate early promoter (a kind gift from Dr. A. Flugel, Department of Neuroimmunology, Max-Planck Institute of Neurobiology, Martinsried, Germany). The propagation and purification of recombinant adenovirus was performed in 911 cells as described previously. 24 In brief, the 911 cells were transduced at a multiplicity of 5–10 and harvested after 36–48 h. The virus was released by five freeze–thaw cycles and purified using two CsCl gradients. The banded virus was recovered, desalted over sephadex columns (Pharmacia, Buckinghamshire, United Kingdom), and stored in virus storage buffer after addition of 10% glycerol at −80°C. Titration of the virus concentration after elution from the column was performed by plaque assay on the 911 cells (plaque-forming units).
MNCs were seeded in 12-well plates at a density of 1 × 107 cells/well in 2 mL of endothelial basal medium-2 (EBM-2) with growth factors. After 7 days, culture medium was removed, and cells were incubated with AdEGFP at different virus/cell ratios (multiplicity of infection [MOI]) of 100, 500, or 1000 in 500 μL medium for 1 h at 37°C. Untreated cells served as control. Then supernatant was removed, and cells were further incubated in fresh medium for 24 h at 37°C. To enhance transduction efficiency, AdEGFP was spun onto the cells by centrifugation for 60 min, at 2000 g and 37°C. Then supernatant was removed, and cells were further incubated in fresh medium for 24 h at 37°C.
Adeno-associated virus
Pseudotyped adeno-associated virus (AAV) vectors (AAV2 genome) were generated using a dual plasmid transfection of HEK 293T cells with a recombinant plasmid containing the GFP transgene within AAV2 ITRs under control of the CMV promoter (pTRUF-2) 25 and the appropriate plasmid for the various serotypes AAV1, 2, 4, 5, and 6,26,27 using established protocols. 28 Briefly, equimolar amounts of plasmid were transfected by calcium phosphate precipitation of 293T cells. After 48 h, cells were harvested and lysed, releasing the virus, followed by treatment with endonuclease (Sigma) to degrade nonencapsulated DNA. The clarified lysate was further purified by density centrifugation in iodixanol (Axis-Shield, Cambridgeshire, United Kingdom), and crude virus was removed and applied to prepared affinity chromatography columns (Heparin or Q-Sepharose columns; Amersham). Purified virus was eluted and concentrated in a centrifugal filter device (Millipore, Cork, Ireland) and stored at −70°C. Titration was performed by real-time PCR of viral samples, and expressed as DNAse-resistant particles (drp)·μL−1, using a probe for the CMV promoter (Operon, Cologne, Germany).
At day 7, medium was aspirated off cultured EPCs and the equivalent of either 5 × 104 or 2 × 105 drp/cell of pseudotyped AAV2/1, AAV2/2, AAV2/4, AAV2/5, or AAV2/6 in 1 mL serum-free medium was added to each well. Plates were centrifuged at 1200 g for 90 min at 34°C and replaced in a 37°C incubator with 5% CO2 and 95% humidity. After 12–16 h, 2 mL complete EPC medium was added, without aspirating off the viral load. Cells were analyzed for GFP expression at 3 days posttransduction.
Lentivirus
Two rHIV-1–based lentiviruses were used in this study. Both were pseudotyped with vesicular stomatitis virus glycoprotein (VSVG) envelope and encoded the GFP gene. PPT-PGK-GFP-WPRE, a third-generation vector containing the PGK promoter, was produced by Genethon, Evry Cedex, France (ref. pG3.22116). pWPT-GFP, a second-generation vector containing the EF1α promoter, was produced in-house using the pWPT-GFP, pRSV-rev, pMD2.G, and pMDLg/pRRE plasmids, which were a kind gift from Didier Trono, Lausanne. Both virus vectors were produced using standard four-plasmid calcium phosphate transfection on 293T cells. Viral titers were determined by transduction levels in HeLa cells and correlated to ELISA for p24 viral protein. EPC culture medium was aspirated off, and cells were transduced at MOI of 1, 10, and 50 in 500 μL of fresh medium before being replaced at 37°C and 5% CO2 for 1 h. After 1 h, 1 mL of fresh medium was applied, and medium was replaced after 24 h. The cells were then maintained in culture for a further 48 h before analysis.
Liposomes
EGFP-C1 plasmid (Clontech, Mountain View, CA), which contains the GFP gene under the control of the CMV immediate-early promoter, was prepared using a Qiagen Endofree™ (Crawley, United Kingdom) plasmid preparation kit, as per manufacturer's instructions.
Liposome complexes were formed using EGFP-C1 plasmid and Lipofectamine 2000™ reagent (Invitrogen, Carlsbad, CA). Before transfection, 10 μL Lipofectamine 2000 reagent was mixed with serum-free medium, and 1 μg plasmid was mixed with serum-free medium. These were incubated for 45 min before forming complexes. When lipid and DNA was mixed, it was left for a further 15 min. Medium was removed from cells, and complexes were mixed in serum-free medium and overlayed on cells. After incubation (6–24 h), complexes were removed and serum-containing medium was placed on cells for a further 48 h. Cells were then harvested, fixed and analyzed using Guava Expressplus assay.
Electroporation
EGFP-C1 plasmid (as described above) was used for the electroporation experiments.
Cells were prepared from whole blood as per standard protocol and plated onto 12-well plates. Nonadherent cells were pipetted off and retained, and adherent cells were harvested from well and added to stock of nonadherent cells. These were spun at 1500 rpm for 5 min.
Cells were resuspended in 400 mL of basal EBM medium containing 20 μg plasmid and placed in 0.4 cm electroporation cuvettes. Cells were electroporated (according to parameters below) and allowed to sit at room temperature for 10 min. Electroporated cells (and nonelectroporated controls) were then plated onto fibronectin-coated 12-well plates, supplemented with EBM (complete) and monitored for expression at 24 and 48 h.
Electroporation was carried out on a Biorad Gene Pulser Electroporator and electroporation parameters were chosen with reference to previously described techniques.29,30
Analysis of gene expression and cytotoxicity
All cytometric analysis was carries out using the Guava Express Plus program on the Guava Easycyte (Guava Technologies, Hayward, CA) except the population separation experiments that were carried out on the FACS Canto flow cytometer (BD, Franklin Lakes, NJ). Cells were harvested by trypsinization, and an equal volume of complete endothelial growth medium-2 was added to stop the reaction. The cells were then centrifuged for 5 min at 300 g, and the pellet was resuspended in 250 μL of phosphate-buffered saline and 4 μL of 7-amino actinomycin D fluorescent dye (Cell Technology, Mountain View, CA). This dye stains apoptotic and dead cells by permeating compromised membranes and binding to DNA. 31 Cells were again washed before fixation in 4% paraformaldehyde (250 μL) at 4°C for 20 min. The cells were centrifuged again as described above and resuspended in 200 μL of cell wash buffer (BD Biosciences, Oxford, United Kingdom) for analysis of GFP expression and 7-amino actinomycin D binding.
Matrigel tubule assay
Matrigel (Sigma) was thawed on ice and placed onto four-chamber glass slides (Nalgene Nunc, Ros Kilde, Denmark) at 37°C for 30 min to allow solidification. About 2 × 104 1,1 dioctadecyl-6,6-dil(4-sulfophenyl)-3,3,3 indocarbocyanine-acctylated-low density lipoprotein (DiI-acLDL)–labeled EPCs (control or transduced with adenovirus at MOI of 500) were cocultured with 4 × 104 human umbilical vein endothelial cells (HUVECs) (passage 3) on the preplated Matrigel. The number of DiI-acLDL EPCs incorporated to the tubule was determined in five random high-power fields in duplicates. A tubule was defined as a structure exhibiting a length four times its width.
Results
EPC culture
Human EPCs (hEPCs) were isolated from patients using a modified protocol as described above. After 7 days in culture, a monolayer of cells exhibiting heterogenous morphology developed on the fibronectin substrate (Fig. 1A). These cells were found to stain positively for uptake of DiI-labeled acetylated LDL and UEA-1 Ulex lectin (Fig. 1B) and were deemed to be EPCs as per convention. 32 Positive staining was also observed on the cell surface for the endothelial marker KDR (Fig. 1C) and the hematopoietic marker CD34 (Fig. 1D).
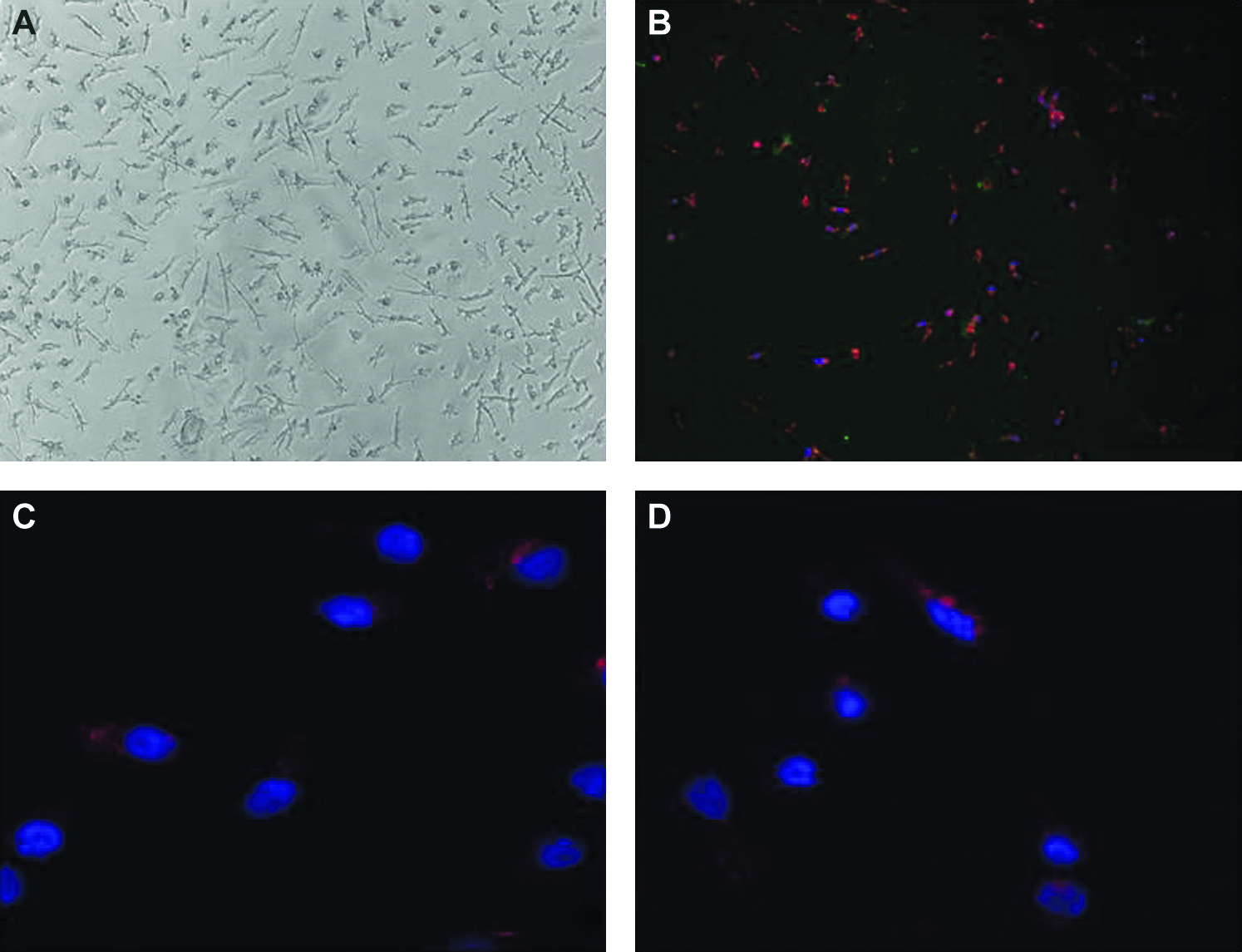
Staining of EPCs. (
Transduction of hEPCs with recombinant adenovirus
hEPCs were isolated and transduced with recombinant adenovirus using different MOI. Of all vectors tested in this study adenovirus proved the most efficient at transducing hEPCs. GFP expression was observed in cells using direct fluorescent microscopy (Fig. 2A) as well as flow cytometry with efficiencies of up to 80% and very low levels of cell death (<1%). Using standard transduction methods, cells expressed GFP at levels up to about 15% (average 12.6 ± 2.3%) at MOI of 1000 with a dose-dependent response relating to different MOI. However, the transduction efficiency was increased fivefold (p < 0.05) with an average of 54.4 (±13.3)% by the incorporation of a centrifugation step that also largely abrogated the dose-dependant response (Fig. 2B). Data represent combined results of experiments carried out in triplicate with n = 3 per experiment for each MOI.
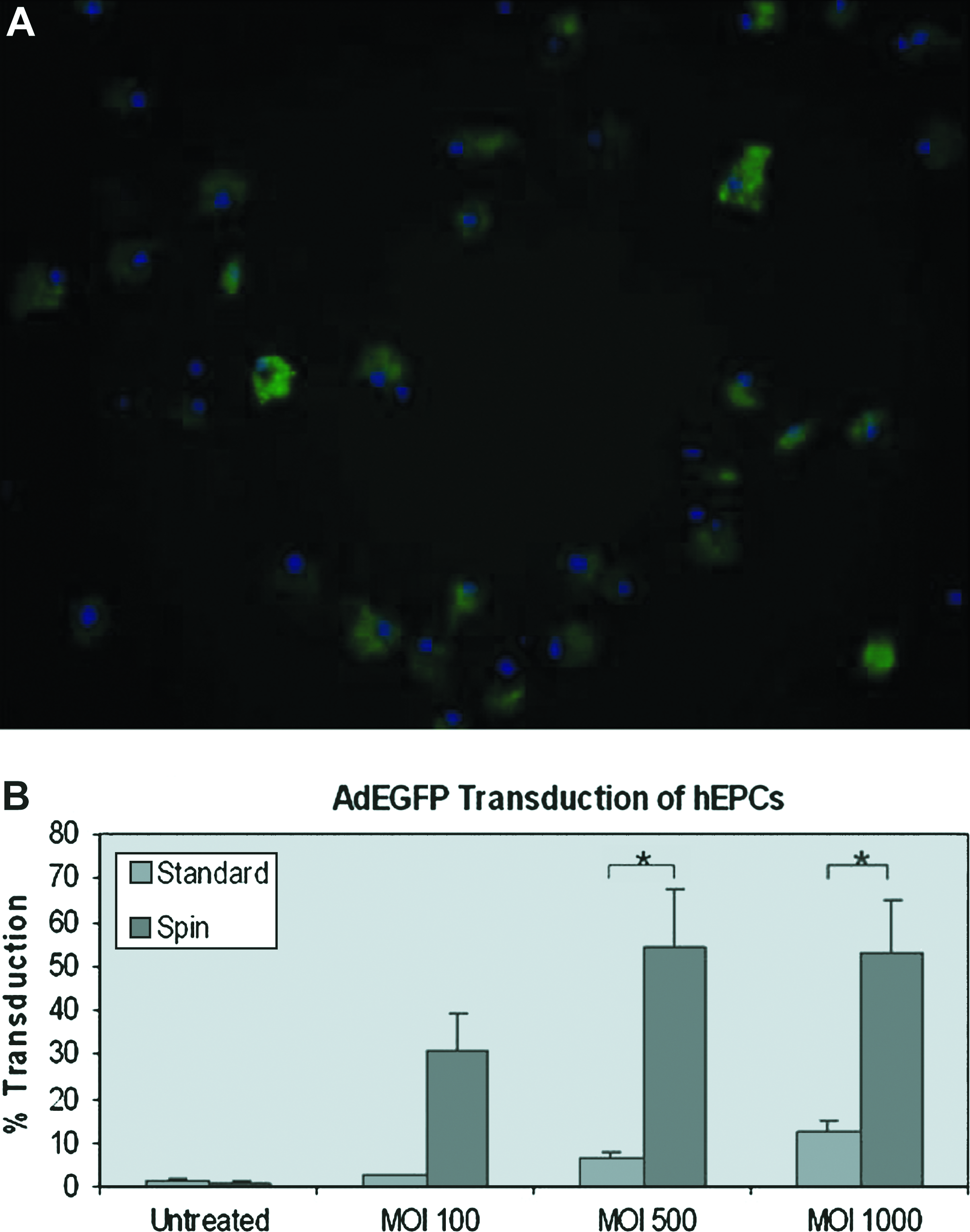
Adenovirus-mediated gene transfer in EPCs. (
Identification of different cell subpopulations in hEPC cultures
Two subpopulations of cells were identified in the original hEPC parent population, separated on the basis of cell size. Transduction efficiencies of these subpopulations were investigated using adenovirus as it proved to be the optimal gene transfer protocol. Of these subpopulations, one population (designated as subpopulation 1 and which comprised about 31 (±2.1)% of the total population) showed remarkable resistance to transduction with adenovirus, with less than 1% of cells expressing the transgene (Fig. 3A), while the second population (subpopulation 2) was very effectively transduced by adenovirus showing up to 87.7 (±2.2)% expression at an MOI of 500 (Fig. 3B) with minimal cell death comparable to subpopulation 1.

Comparison of transduction efficiencies for two separate subpopulations in parent EPC population after cytometric sorting based on cell size. (
Transduction of hEPCs with AAV vectors
hEPCs were isolated and transduced with recombinant AAV using different drp. Surprisingly, we observed substantial variation in AAV efficiencies between repeated experiments with this pattern being displayed by all serotypes tested. However, this variation much more pronounced over experiments involving different donors. Intraexperimental variation, where three samples were generated from the one donor, was greatly reduced in comparison. The greatest variation observed was in AAV2/1 experiments with interexperimental standard error being as high as 10.5%. However, over three different experiments the average intraexperimental standard error was much lower at 3.3 (±0.3)%. Overall, the highest expression obtained for this vector was using AAV serotype 2 with efficiency up to a maximum of 38.89% with the average over three series of experiments (n = 3 for each experiment) being 25.32 (±7.29)% with drp of 200,000. Increases in drp from 50,000 to 200,000 did not show proportional increases in GFP expression levels although some increase was seen with serotypes 2, 5, and 6 (5.68%, 5.86%, and 8.09%, respectively) (Fig. 4A).

Gene transfer in EPCs using AAV-, Lenti- and nonviral vectors. (
Transduction of hEPCs with recombinant lentiviral vectors
hEPCs were isolated and transduced with recombinant HIV vectors using different MOI. The HIV-1–based, VSVG-pseudotyped lentiviruses used in this study showed relatively low levels of transduction with maximum expression at an MOI of 50 and little improvement at higher levels. Using a PGK promoter driven construct, a dose-dependent response was seen with increasing virus titers of 1, 10, and 50 with cell death levels being minimal (<1% due to exposure to vector). The maximum expression seen in a single experiment was 36.9% with the average result from two different experiments being 25.8 ± (11.1)%. Here, again, substantial variation was seen over the entire series of experiments. At an MOI of 50, standard error was as high as 11.1% across three different experiments, while the average standard error within these experiments was 2.28% (Fig. 4B). Using the EF1α promoter, almost no gene expression was measurable, this result being confirmed by direct fluorescence microscopy. However, in HeLa cells transduced with this virus, efficiencies of up to 100% were observed at an MOI of 10 (data not shown).
Transduction of hEPCs using plasmid–DNA/liposome complexes
Liposomal gene delivery reliably transfected cells although at low levels. Expression was evident in 29.2 (±1.1)% of cells after 6 h with similar levels being observed after 24 h (Fig. 4C). Uptake of plasmid alone was negligible when compared to levels achieved with the use of Lipofectamine 2000. Experiments were performed in triplicate with n = 3 per experiment.
Electroporation
Electrotransfer techniques were found to be unsuitable for the cell populations studied. Increasing voltages led to high cell death with very little transgene expression observed in any remaining viable cells (data not shown).
Endothelial tubule formation
When examined for ability to incorporate into endothelial tubules, there was no statistical difference between the mean number of incorporated EPCs in control group and group transduced with adenovirus at MOI of 500 (mean ± SEM, 10.9 ± 0.6 vs. 11.1 ± 1.0; p = 0.87), suggesting that the transduced EPCs did not exhibit in vitro functional impairment (Fig. 5A). Figure 5B shows GFP-positive EPCs incorporating into endothelial tubules after coculture with HUVECs at a ratio of 1:2.

EPC tubule formation in Matrigel. (
Discussion
The homing abilities of stem cells have made them attractive candidates to carry therapeutic genes after genetic modification in the field of gene therapy, and embryonic, 33 hematopoietic, 34 and mesenchymal 35 stem cells have all been considered. EPCs have been found to have the ability to home directly to areas of endothelial and vascular damage and neoangiogenesis, 36 making them particularly attractive for use as agents to deliver gene products after being genetically modified when only selected areas are to be targeted.
While there have been studies carried out to investigate the use of EPCs overexpressing a transgene of interest,11,12,14,37 there has not, to our knowledge, been a direct comparison of different vectors to determine the most effective method of genetic modification of these cells. In this study, we have compared both viral and nonviral gene delivery systems for efficiency in delivering the GFP reporter gene to cells cultured under identical conditions. Different transduction protocols have been used for different vectors based on preliminary studies to determine the optimum MOI ranges and time-points for analysis for each vector used. These protocols differ owing to the fact that different MOI are required to achieve similar transduction levels when using different vectors in a particular cell type. Indeed, with lentivirus, increased viral titer may have cytotoxic effects on the target cells, mediated by the VSVG envelope.38,39
From the results obtained, adenovirus seems to be the most efficient vector with efficiencies as high as 80% observed. Previous reports have suggested the need for the use of high MOI (up to 1000), 14 but these results are consistent with results we obtained using a standard transduction procedure in which a dose-dependent response was seen with efficiency levels only rising to about 15% at high MOI (1000). However, when a centrifugation step was introduced, efficiency levels rose dramatically averaging about 50% across a series of experiments with the dose-dependent response not being seen above an MOI of 500, and with minimal levels of cell death. It is possible that the use of the centrifugation step allows a physical proximity between virus and cell that bypasses the reliance on high levels of coxsackie virus and adenovirus receptor (CAR) receptor expression on the EPC. This receptor is used by the virus for loose binding to the cell surface, 40 which is provided for by the physical force of the centrifugation, thus making the adenovirus vector more reliant on the integrin family of cell surface heterodimers 41 for internalization. However, this is speculative as we are not aware of any study examining the expression levels of CAR on EPCs. In addition, the low proliferative rate of these cells negates one of the inherent disadvantages of using adenoviral-mediated genetic modification which is that there may be a diluting out effect in other more proliferative cell types. Early EPCs tend to die off after 7 days and modification is likely to be for the life of the cell. Regarding modification of rapidly dividing outgrowth EPCs, this is of great importance, and use of a vector such as lentivirus that can incorporate the gene of interest into the genome may be appropriate. 42
Lentivirus has also been shown to efficiently transduce nondividing cells, 43 which makes it seem an attractive candidate for gene transfer to low proliferative, early EPCs. However, our results have shown lower transduction efficiencies using this vector than with adenovirus. We have shown transduction levels of up to 37% at an MOI of 50 with minimal cell death due to the virus seen. However, overall efficiency of lentivirus transduction was inconsistent with fluctuating levels between different experiments and overall levels being in the region of 25% at an MOI of 50. In contrast to the study published by Liu et al., we observed a higher efficiency of transduction using the PGK promoter than with the EF1α promoter. 44 In this study, a centrifugation step was included in the transduction protocol that may account for the higher overall gene expression rates observed. We did not include such a step because prior experiments with rat mesenchymal stem cells showed that this technique conferred no advantage with lentiviral vectors (data not shown). This raises interesting questions concerning the method of entry of lentivirus vectors to different cell lines from different species.
AAV has many advantages for gene transfer such as absence of vector toxicity and prolonged transgene expression. AAV-mediated gene delivery to EPCs showed a large degree of variation in a series of experiments, with AAV2/2 showing the highest levels at around 25%. An increase in drp made little or no difference with serotypes 1, 2, and 4. In addition, we observed a significant degree of variability in interdonor transduction efficiency using this vector. In repeat experiments using cells from the same donor, efficiency of transgene expression was more consistent. Although a degree of species specificity has been shown using AAV, 35 the reason for this variation using cells from different donors is unclear. It is possible that it is due to low receptor expression as the same degree of variation was not seen with all serotypes tested. Indeed, it has been suggested that low levels of receptor expression account for variability observed between different donors in experiments to transduce hematopoietic progenitor cells. 45
Although viral vectors are traditionally regarded as being more effective than nonviral vectors, we found comparable transgene expression efficiency in cells transfected using lipofectamine for 6 h to those transduced with lentivirus or AAV. Liposome-mediated gene delivery to EPCs resulted in transgene expression in up to 35% of cells after 24 h accompanied by low rates of cell death. In future studies, the use of nucleofectin could be explored to improve efficacy.
Enhancement of plasmid uptake by electroporation proved unsuitable for this cell type. Minimal gene transfer was observed at the parameters tested with high levels of cell death. Membrane pore formation is influenced by the radius of curvature of a cell, and more asymmetric cells have been found to be more amenable to permeabilization than more regular cells. 46 EPCs tend to attach strongly to a substrate such as fibronectin when in culture, allowing them to develop varied morphologies. However, when detached from the surface on which they have been growing, they adopt a much more uniform and rounded appearance. This may affect the ease with which the membrane can be permeabilized by an electric field with the voltages required to porate them being too high for the cell to tolerate. In addition, it has been shown that cells derived from a monocytic lineage were difficult to electroporate. 47 EPCs are also suspected to be of monocytic origin as evidenced by their expression of CD14. 48 These monocytic characteristics may also go some way toward explaining the relatively low levels of cell death observed when using viral vectors and liposome-mediated gene transfer. If these cells are from a lineage concerned with defense in vivo, a certain level of robustness could be expected.
In terms of functionality, exposure of the cells to adenovirus appears to have little or no effect on ability of cells to incorporate into a 3D endothelial structure as seen after coculture of transduced EPCs with mature endothelial cells growing in Matrigel. No difference was observed in levels of incorporation over untreated controls after transduction with the most efficient vector identified in this study. This may have implications in vivo, but this was not within the scope of the current study.
Basic morphology observed in a monolayer of cultured early EPCs suggests a heterogeneous population. However, no reliable markers have so far been identified to separate out different populations. In this study, we have identified at least two different populations that we have separated by flow cytometry based on cell size. Morphologically, a spindle-shaped cell type and a more cuboid cell type may be observed in the parent EPC population, but whether these different types correspond to the different sized populations seen with flow analysis is unclear. Because adenovirus had been found to be the most efficient vector for EPC transduction, it was used to transduce both populations using the same protocol as previously described, and we noted a marked difference in their transduction efficiencies. It is clear from our data that one population, making up about 70% of the total population, is responsible for almost all observed expression of the transgene, while the second population does not appear to be easily transducible. In the absence of a specific marker to facilitate separation of these populations, the differing transduction efficiencies may provide a means by which they may be separated for further and individual analysis. These findings raise interesting questions concerning the relative characteristics of these different subpopulations and the differing transduction potentials, but this was not the focus of the current study.
In summary, this study has shown, by comparison of both viral and nonviral vectors, that human early EPCs are most readily transduced by adenovirus. We have also shown that transduction and reporter gene expression do not have a detrimental effect on the ability of these cells to incorporate into an endothelial monolayer. Further, we have identified two distinct populations within the heterogenous parent population that show markedly different capacities for adenoviral transduction. This provides a useful procedure to facilitate the use of this cell type in conjunction with gene therapy in a range of studies.
Footnotes
Acknowledgments
The authors gratefully acknowledge the assistance of Margaret Scarry, Angela Moore, Mary Bohan-Keane, Keara O'Hehir, Sinead Syron, Mary Manning, and all the staff of the hemochromatosis clinic at University College Hospital Galway and that of Lisa McGinley in virus preparation.
This work was funded by Irish Research Council for Science, Engineering, and Technology (IRCSET), and by Science Foundation Ireland (SFI) Centre for Science Engineering and Technology (CSET) awards.
Disclosure Statement
No competing financial interests exist.