
Abstract
The influence of an acellular porcine matrix on proinflammatory activation of endothelial cells (EC) during normoxia and hypoxia was investigated by a newly established semi-quantitative electron microscopic procedure. As a model, three adhesion molecules (E-selectin, ICAM-1, and VCAM-1) were localized by silver-enhanced immunogold staining and energy dispersive X-ray microanalysis after normoxic or hypoxic pretreatment of the cells and subsequent stimulation with IL-1β. Morphology of EC grown on porcine matrix or coverslips was recorded simultaneously using secondary electron imaging. EC appeared tightly attached to the underlying surfaces with their typical cobblestone-like morphology. Statistically significant upregulations upon stimulation with IL-1β were observed in both groups for all three adhesion molecules. Hypoxic pretreatment of the specimens with subsequent reoxygenation neither induced morphological changes nor caused an upregulation of adhesion molecule expression in cells grown on acellular porcine tissue. Unexpectedly, in cells seeded onto the acellular matrix, IL-1β failed to upregulate ICAM-1 expression after a short period of hypoxia. The surface expression of VCAM-1 was also significantly lower even under normoxic conditions, which might indicate the development of functional impairment of cells in contact with acellular porcine tissue. The method presented in this study has proven valuable for the determination of antigen expression on scaffold materials in parallel with the characterization of surface morphology.
Introduction
Several different techniques have been shown useful for studying cellular reactions toward exogenous stimuli and for analyzing changes of cellular surface structures. The most important of them are ELISA, flow cytometry,1,2 scanning electron microscopy (SEM), laser scanning confocal microscopy, 3 and polymerase chain reaction. 4 So far, their value is limited by one major shortcoming: the incapability of providing data on immunological changes, for example, altered expression of cellular surface antigens simultaneously with the delivery of morphologic information at high resolution. Identification of the local distribution of immunological surface markers as well as imaging of structural alterations of cells and scaffold materials are prerequisites in the field of tissue engineering.
The present study aims at developing an in vitro method capable of delivering detailed information on cell morphology in parallel with a quantification of cell surface molecules of EC grown on acellular scaffolds at baseline and during activation. A well-investigated technique for visualization of surface receptors in SEM is the immunogold staining technique, which uses colloidal gold particles conjugated to specific antibodies as markers. 5 The silver enhancement technique provides specific enlargement of the bound gold particles by precipitation of metallic silver, thus permitting the recognition of bound antigen on SEM micrographs.
Energy dispersive X-ray analysis (EDX),6–8 depending on the specific X-ray spectrum emitted by elemental silver, allows automated identification of silver particles on the cell surface and digital storage in dot maps. Digital image analysis of these maps provides semi-quantitative information on antigen expression in cells during contact with a biomaterial surface.
This new combination of techniques provides comprehensive information on surface morphology and immune activation simultaneously and might therefore be expected to become a valuable tool for addressing a variety of experimental questions.
Materials and Methods
Porcine pulmonary valve conduits (n = 8) consisting of the semilunar leaflets and the pulmonary artery truncus were obtained from a local slaughterhouse, and were transported and incubated for 24 h in RPMI 1640 medium containing multiple antibiotics for sterilization. 9
Decellularization procedure
Porcine heart valve conduits were decellularized according to a protocol that was developed in our laboratory.9,10 In brief, the conduits were treated with 0.05% Triton-X 100® (Bio-Rad, Hercules, CA), 0.05% sodium deoxycholate (Merck, Darmstadt, Germany), and 0.05% Igepal CA-630® (ICN Biomedicals, Aurora, OH) under permanent shaking for 48 h at 4°C. Subsequently, the conduits were incubated with 100 μg/mL ribonuclease (Roche Diagnostics GmbH, Mannheim, Germany) and 150 IU/mL deoxyribonuclease (Sigma, St. Louis, MO) with 50 mmol/L MgCl2 in Dulbecco's phosphate-buffered saline (D-PBS; Invitrogen, Paisley, Scotland, United Kingdom) under permanent rotation for 24 h at 37°C. The samples were washed thoroughly with M-199 medium (Gibco, Paisley, Scotland, United Kingdom) and 0.2% EDTA (Merck) under permanent shaking for 5 days at 4°C and again washed with M-199 medium alone under permanent shaking for 5 days at 4°C. Thereafter, decellularized conduits were cryopreserved in sterile tubes containing RPMI 1640 medium and 10% dimethyl sulfoxide. Cryopreservation was carried out in an automatic freezing device (Ice Cube 1610 Computer Freezer; SY-LAB, Neupurkersdorf, Austria) as outlined in Seebacher et al., 11 and conduits were stored at −80°C until use.
Endothelial cell culture
Human umbilical vein endothelial cells (HUVEC) were purchased from Promocell (Heidelberg, Germany) and cultured in M199 medium (pH 7.4) containing 20% fetal calf serum (FCS), 100,000 U/L penicillin (Gibco), 100,000 μg/L streptomycin (Gibco), 100,000 U/L low molecular weight heparin (Sigma, Munich, Germany), and 30 mg/L bovine hypothalamic growth factor (Upstate Biotechnology, Lake Placid, NY). The confluent primary and single donor HUVEC monolayers (n = 10) were washed with D-PBS (Gibco), trypsinized, harvested, centrifuged for 5 min at 300 g, and subcultured in M-199 medium with the same supplements as above. Only cells from these first subcultures were used for the experiments described below. Cells were identified as EC by the typical cobblestone-like morphology and by factor VIII (vWF) staining.
Endothelial cell seeding
HUVEC (4 × 104 cells/cm2) were either seeded onto cell culture–treated Thermanox™ plastic coverslips (n = 48) (Nunc, Rochester, NY) precoated with a 1% solution of bovine gelatin (Sigma, St. Louis, MO) or onto pieces of the decellularized porcine heart valve matrix (n = 45) and kept under static conditions in a humidified incubator (37°C, 5% CO2) until confluence was reached. To better illustrate the number of protein attachment per single cell, short-term studies were performed. For this purpose HUVEC (1 × 104 cells/cm2) were seeded onto pieces of the decellularized porcine heart valve matrix (n = 12) and kept under static conditions in a humidified incubator (37°C, 5% CO2) for 24 h.
Hypoxia and reoxygenation
CO2-independent medium (Gibco) containing 10% FCS was degassed by streaming with pure nitrogen. Cell culture medium was removed and replaced by degassed CO2-independent medium. Seeded specimens were kept in cell culture plates in a chamber purged of oxygen by 100% nitrogen for 60 min (total n = 41; porcine tissue n = 20 and coverslip n = 21), while the other group was kept at room air conditions (total n = 46; porcine tissue n = 22 and coverslip n = 24). All test specimens were kept at 37°C. At the end of the incubation, samples were transferred into a humidified incubator (37°C, 5% CO2). To stimulate adhesion molecule expression, one part of the specimens in each group (normoxia vs. hypoxia) was incubated with IL-1β (10 ng/mL) (R&D Systems, Minneapolis, MN) for 4 h (E-selectin), 20 h (ICAM-1), or 48 h (VCAM-1). Specimens without IL-1β treatment served as unstimulated controls (porcine tissue n = 10 and coverslip n = 12).
In the short-term group (normoxia only), HUVEC grown on porcine heart valve specimens were incubated with IL-1β as described above (n = 6). Specimens without IL-1β treatment served as unstimulated controls (n = 6).
At the end of the incubations, supernatants were withdrawn, and samples were washed with D-PBS and prefixed with 0.1% glutaraldehyde (15 min, 4°C). After thorough washing, cells were incubated with a solution of 0.05 mol/L glycine (Merck ) in D-PBS (15 min, 37°C).
Gold labeling of cells and silver enhancement
Thorough washing (1% bovine serum albumin [ICN Biomedicals] and 1% goat serum [Sigma, St. Louis, MO] in D-PBS = buffer A) was followed by incubation (60 min, 37°C) with monoclonal mouse anti-human antibodies recognizing E-selectin, ICAM-1, or VCAM-1, each in a solution of 5% bovine serum albumin and 4% goat serum (=buffer B) (E-selectin [CD62E]: clone BBIG-E4[5D11] and concentration 2 μg/mL; ICAM-1 [CD54]: clone BBIG-I1[11C81] and concentration 2.5 μg/mL; VCAM-1 [CD106]: clone BBIG-V1[4B2] and concentration 5 μg/mL; all from R&D-Systems). Repeated washing with buffer A was followed by incubation with a gold-conjugated goat anti-mouse secondary antibody (5 nm particle size; AuroProbe™; Amersham Biosciences, Buckinghamshire, United Kingdom) for further 60 min (37°C). Again, samples were washed twice (buffer A), fixed with 2% glutaraldehyde for at least 20 min at room temperature, and finally washed thoroughly with bidistilled water. Subsequently, silver enhancement was performed with the IntenSE™ M-kit (Amersham, Uppsala, Sweden). 12 The formation of nonspecific silver precipitates (auto-nucleation) was avoided by strictly adhering to the incubation time recommended by the manufacturer. To validate the incubation time for silver enhancement, time-course experiments were conducted in our laboratory, which confirmed the information given in the instruction manual. Negative controls were run in parallel omitting the primary antibody. Closing thorough washing with bidistilled water completed the staining procedure. After graded dehydration with ethanol solutions, specimens were CO2 critical-point dried and ultimately sputter coated with gold–palladium.
Scanning electron microscopy, energy dispersive X-ray analysis (EDX), and quantitation
Secondary electron imaging (SEI) and EDX were carried out using a JSM-5400 scanning electron microscope (Jeol Ltd., Tokyo, Japan) fitted with a LINK Pentafet SiLi detector (Oxford Instruments, Buckinghamshire, United Kingdom), interfaced to a LINK ISIS analyzer (Oxford Instruments).
All analyses were performed with an accelerating voltage of 15 kV and a take-off angle of 0°. Detector–specimen distance was 15 mm, and a constant magnification of 350 × and filament load-current of 50 μA were set for EDX.
Before each EDX session, energy calibration to a manganese standard was performed. Process time for X-ray acquisition was set to 1 (high acquisition rate) and a noise limit of 100 μs was defined to suppress random background signals, frequently appearing in dot maps. 13
For qualitative detection of silver in the LINK ISIS analyzer, an energy spectrum of a silver-enhanced specimen was recorded and stored. Energy peaks representing gold and silver were identified using the auto-ID function of the ISIS LINK software. The energy window was set to the X-ray emission peak from the Lα1-shell of silver (2.9844 keV). Data acquisition on the scanning electron microscope was performed in PHOTO mode (86.4 s/frame) at a resolution of 1908 × 1477 pixels. SEI recording and X-ray area mapping were achieved with a SemAfore™ slow-scan digitizer (version 5.01; Jeol Skandinaviska AB, Sollentuna, Sweden).
X-ray maps were analyzed and quantitated using the public domain image-analysis software ImageJ 14 running on a standard PC. Five randomly selected sections were defined in each of the thresholded images. Dots representing silver particles in the defined regions were quantitated using the “analyse particle” function of the ImageJ software.
Statistical analysis
Particle count, area fraction bearing particles, and particle size were compared between treatment groups using the SPSS software (version 11.5). Data are given as mean particle count ± SD. Statistical significance was calculated by means of the paired t-test. Statistical significance was set at p < 0.05. For multiple comparisons a Bonferroni correction was applied.
Results
Evaluation of the SEM micrographs revealed that EC were tightly attached to the acellular porcine matrix or coverslips, respectively. The typical cobblestone-like morphology of confluent EC monolayers is depicted in Figure 1A and 1B. The preparation method used for the specimens proved to be adequate for the preservation of cellular structures during SEM examination. Small globules on the EC surface of stimulated samples representing the silver-enhanced antibodies could be easily recognized (Fig. 2B). Short-term studies with HUVEC grown on the acellular porcine matrix and subsequent SEM examination at high magnification (2000 ×) allowed the visualization of protein attachment on single cell level (Fig. 3A, B). During EDX analysis the constant counting rate of all X-ray lines produced by silver allowed for reliable determination of the immunochemical reaction product. The accelerating voltage of 15 kV permitted the accurate subtraction of background and the characteristic silver peaks from the recorded spectrum. A control group with omission of the primary antibody produced only a marginal background signal (particle count: 111 ± 16, mean ± SD, n = 5).

SEM micrographs (350 ×). (

Exemplary process sequence. (
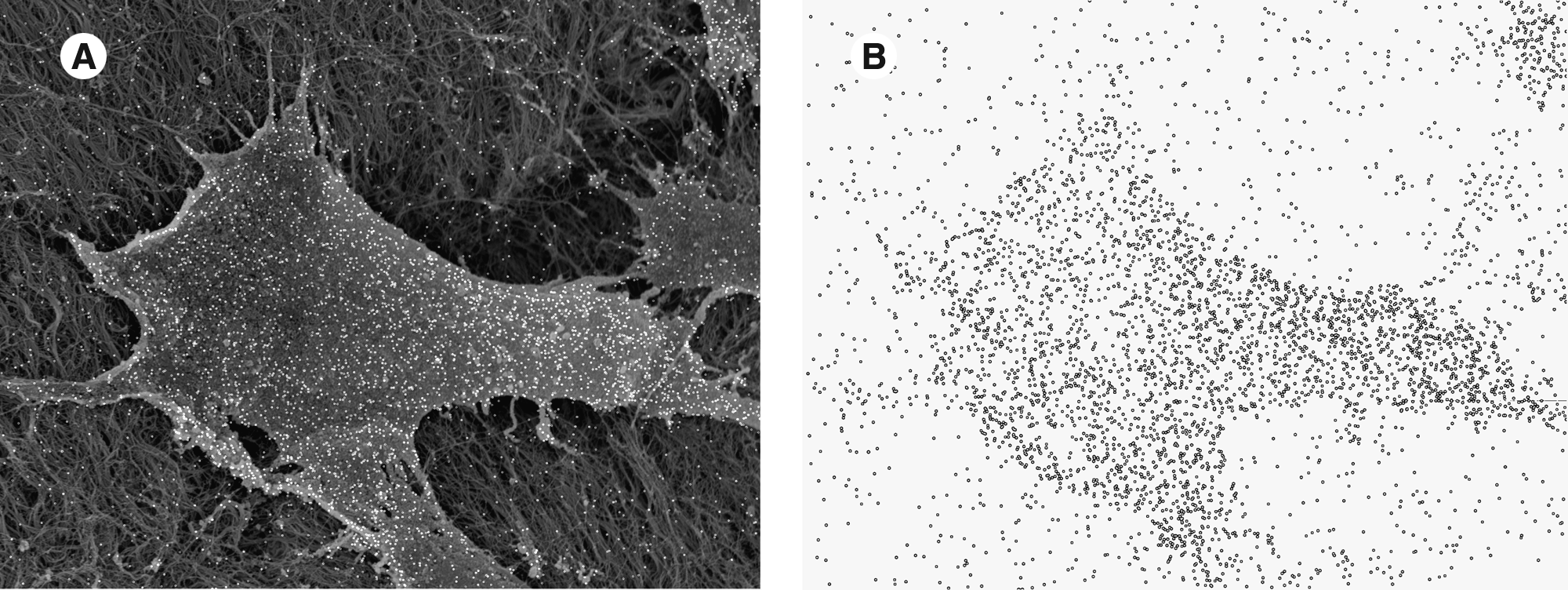
(
E-selectin
Under both normoxic and hypoxic conditions, IL-1β induced a statistically significant rise in E-selectin expression when cells were grown on coverslips (p < 0.01, vs. unstimulated samples). Seeding of EC onto acellular porcine tissue had no adverse effect on the responsiveness toward activation with IL-1β (p < 0.01, vs. unstimulated samples) when cells were kept under normoxic conditions. In contrast to coverslip controls, the IL-1β–induced increase in E-selectin expression was less pronounced when cells had been exposed to a period of hypoxia (coverslips, 1060 ± 95 particles; porcine matrix, 841 ± 245 particles; p < 0.01) (Fig. 4A).

Bar graphs showing the adhesion molecule expression of HUVEC seeded onto acellular porcine tissue or plastic coverslips. E-selectin (

ICAM-1
Under normoxic conditions, a constitutive expression of ICAM-1 was detected for EC grown on coverslips. Treatment with IL-1β induced a statistically significant increase compared to nonstimulated samples (p < 0.01). When HUVEC were grown on acellular porcine tissue, a constitutive expression of ICAM-1, as it was observed for coverslip samples, was recorded. Again, stimulation with IL-1β led to a significant upregulation of endothelial ICAM-1 (p < 0.01, compared to nonstimulated samples). Hypoxic pretreatment of cells grown on coverslips or porcine matrix did not influence constitutive ICAM-1 expression. When cells were grown on the acellular porcine matrix and exposed to a short (60 min) period of hypoxia, the responsiveness of the cells toward IL-1β stimulation was abolished (Fig. 4B).
VCAM-1
IL-1β–induced activation of EC grown on coverslips led to a statistically significant increase of VCAM-1 expression (p < 0.01 compared to nonstimulated samples). Sixty minutes of hypoxic incubation and subsequent reoxygenation led to a slight but statistically significant enhancement of VCAM-1 expression in cells grown on coverslips (p < 0.05 vs. normoxic coverslip sample). Hypoxic pretreatment and subsequent stimulation with IL-1β did not further upregulate VCAM-1 in cells grown on coverslips. Interestingly, cells grown on porcine decellularized specimens displayed a considerably lower VCAM-1 expression upon stimulation with IL-1β when compared to HUVEC grown on coverslips. This effect was seen during normoxic as well as hypoxic conditions (p < 0.01 for both) and might indicate a time-dependent development of functional impairment in EC seeded onto acellular porcine tissue (Fig. 4C).
Discussion
The development of new materials for tissue-engineering purposes is highly dependent on the availability of efficient and time-saving laboratory techniques for the investigation of cell–material interactions. Techniques currently available are mostly restricted to the analysis of either sample morphology or quantitation of cell surface molecule expression. Conventional SEM is restricted to morphological evaluation, whereas laser scanning confocal microscopy provides morphological information and details on the local distribution of CAMs on sectioned specimens only. Major disadvantages of some of the analytical methods for studying cell function are requirements concerning sample preparation with the risk of cell damage or result corruption: flow cytometry requires mechanical or enzymatic cell detachment, while cell ELISA is not compatible with subsequent morphological evaluation.
Several studies used a combination of the mentioned techniques to obtain morphologic and immunological information.15–17 But the interpretation of data obtained by combining results from different analytical procedures bears a considerable risk of inaccuracy.
The principal aim of this study was therefore to develop a method for the simultaneous detection of morphological changes and the semi-quantitative analysis of immunochemical reaction products on the surface of cells growing on scaffolds intended for use in tissue-engineering applications. A combination of immunogold staining with SEM, quantitative X-ray microanalysis, and digital image analysis to gain comprehensive information about morphological and immunological changes on 3D test specimens was established. Silver enhancement was introduced to allow highly sensitive antigenic binding with small-diameter gold-conjugated antibodies and subsequent enlargement of the gold particles by precipitation of metallic silver.18–21
Recording SEM micrographs of the silver-enhanced samples in SEI mode (secondary electron imaging) provides detailed morphologic information of the surface structure and reveals the local distribution of antigenic sites without the need for time consuming overlay of SEI and backscattered electron imaging micrographs presented by deHarven et al. 22 As backscattered electron imaging is based on atomic number contrast, it displays silver distribution but hardly provides sufficient information on surface morphology. In contrast, the identification of elemental silver by defining its X-ray emission peak followed by X-ray mapping, as was used in our study, provides dot maps that represent the surface distribution of bound antibody.
A foreign matrix might lead to phenotypical alterations of EC after seeding. The second aim of the study was to analyze the expression levels of the adhesion molecules E-selectin, ICAM-1, and VCAM-1 at baseline as well as during receptor-mediated stimulation with IL-1β by applying the newly established method described above. Adhesion molecules are very sensitive early markers of proinflammatory activation of the resting endothelium and seem therefore appropriate for the investigation of EC function on a foreign matrix during environmental changes. E-selectin expression on the cellular surface, induced by IL-1, TNF, or LPS, mediates a loose leukocyte adhesion (“rolling”) to the endothelium, while ICAM-1 and VCAM-1 (induced by IL-1 and TNF ) take part in further tight leukocyte adhesion and the following extravasation of leukocytes toward the perivascular space. 23 Leukocyte recruitment to biomaterials largely depends on the expression of adhesion molecules by EC grown on the (bio)artificial scaffold. The initiation of a proinflammatory response is determined not solely by the EC–biomaterial interface but also by environmental conditions cells are exposed to during the surgical procedure (circulating cytokines, hypoxia, and reoxygenation). To predict the contribution of EC adhesion molecule expression to the development of an inflammatory response toward an acellular xenogeneic scaffold in vitro, we simulated an in vivo situation where EC might be exposed to a period of hypoxia during implantation followed by a period of reoxygenation with elevated levels of proinflammatory cytokines.
Examination of the SEM micrographs allowed verification of a mostly confluent monolayer of EC tightly attached to the xenogeneic scaffolds, presenting with their typical cobblestone-like morphology. Semi-quantitative assessment of endothelial adhesion molecule expression using energy-dispersive X-ray microanalysis and digital image analysis clearly showed that the acellular porcine matrix did not induce a proinflammatory activation of EC. At baseline, cells grown on the acellular porcine matrix exhibited only minimal expression levels of E-selectin, ICAM-1, and VCAM-1 that were comparable to those of the negative control (coverslip) group. Upon stimulation with IL-1β, adhesion molecule expression increased in a typical time-dependent course with an initial rise of E-selectin expression, which was followed by increased levels of ICAM-1 and VCAM-1. SEM examination of the silver-enhanced samples in SEI mode (secondary electron imaging) after a 1 h period of hypoxia and subsequent reoxygenation for further 60 min showed a preservation of the surface morphology of cells grown on either porcine tissue or plastic coverslips. Although the acellular porcine matrix did not adversely affect IL-1β–stimulated E-selectin expression, IL-1β–induced upregulation of ICAM-1 following hypoxic pretreatment as well as stimulation of VCAM-1 during normoxia and hypoxia was impaired in EC grown on porcine tissue. This observation is not indicative for the development of a proinflammatory endothelial phenotype, but is a strong hint for a functional dysregulation of cells in contact with a xenogeneic matrix.
In conclusion, the combination of silver-enhanced immunogold staining with SEM, quantitative X-ray microanalysis, and digital image analysis described herein seems to be a valuable tool in tissue engineering research where the distribution of surface antigens on cells in association with the biomaterial surface structure needs to be evaluated. Further potential applications for this novel technique would be the in vitro evaluation of cell function on synthetic graft materials prior implantation, the simultaneous morphological and immunological characterization of adherent cells after mechanical testing of constructs, or the functional characterization of cells on tissue samples harvested during surgery. One shortcoming of the procedure presented in this study is that it does not allow the simultaneous staining of different antigens, and also is limited to the detection of antigens expressed on the cell surface.
Footnotes
Disclosure Statement
No competing financial interests exist.