
Abstract
In tissue engineering, coculture systems have been employed for two major purposes: (1) construction of tissue and organ substitutes (e.g., coculture of parenchymal and nonparenchymal cells in liver tissue engineering) and (2) maintenance of cellular functions (e.g., coculture of embryonic stem cells with embryonic fibroblasts as the feeder cells). For the characterization and recovery of specific cell types, however, target cells have to be isolated from other cells. We report here a novel magnetic separation method to isolate target cells in coculture systems. In this method, target cells were cocultured with nontarget cells labeled with magnetite cationic liposomes (MCLs). Thus, when necessary, the MCL-labeled nontarget cells were magnetically removed from the coculture, resulting in negative isolation of the target cells. As the separation models, three deferent types of coculture systems were examined: rat hepatocytes with various MCL-labeled cells (mouse NIH3T3, STO, or human umbilical vein endothelial cells), human keratinocyte HaCaT cells with MCL-labeled NIH3T3 cells, and mouse embryonic stem cells with MCL-labeled STO cells. In these cocultures, target cells were separated with 94% purity and 98% recovery yield on average. This technique provides a promising approach to isolate and recover target cells for further analysis and application.
Introduction
The drawback to these coculture systems is that for characterization and recovery of specific cell types, the target cells have to be isolated from the other cells. The specific cell separation methods generally used are primarily classified into two categories: fluorescence-activated cell sorting (FACS) and magnetic cell sorting (MCS). Although FACS is a powerful tool to obtain highly purified cells, this method is time consuming for processing large number of cells and requires a special device. On the other hand, MCS is now widely used in research and clinical applications19–21 due to its simplicity. The magnetic particles most frequently used for cell separation are ferrites with the general composition of MFe2O3 (where M represents a bivalent metal cation such as Ni, Co, Mg, or Zn and includes magnetite Fe3O4) and maghemite Fe2O3. For example, surface modification of magnetic particles with specific antibodies allows target cells to attach to the magnetic particles. Thus, the target cells can be magnetically captured and recovered directly from a complex cell suspension such as blood, 22 bone marrow, 23 and peritoneal exudate. 24 However, the immunomagnetic cell sorting system requires the surface antigens to be present on target cells and the specific antibody, which may limit its application.
Functionalized magnetite nanoparticles have been developed by applying the concepts involved in drug delivery systems. 25 Magnetite cationic liposomes (MCLs), which are cationic liposomes containing 10-nm magnetite nanoparticles, exhibited improved accumulation of magnetite nanoparticles in cells by using the electrostatic interactions between MCLs and the cell membrane. 26 Because the cell surface is generally negatively charged, various types of cells can be labeled with MCLs. Based on the fact that MCL-labeled cells can be manipulated using a magnet, this cell manipulation technique has been applied in tissue engineering.27,28 Although the general technology of these processes has been established for use in tissue engineering, there is still room for improvement.
From the point of view of bioprocess engineering, development of a methodology for the physical manipulation of target cells is essential for tissue engineering in the coming decades. We describe here a new tissue engineering technique for the magnetic separation of target cells in coculture systems using MCLs. Generally, in coculture systems the target cells (cells of interest) and the cocultured cells (nontargeted cells, including feeder cells) are often prepared separately. In the present study, the target cells were cocultured with the nontarget cells labeled with MCLs. When necessary, the MCL-labeled nontarget cells were magnetically removed from the coculture, resulting in the negative isolation of the target cells. Feasibility of this strategy was examined using three model coculture systems of hepatocytes with various cells, keratinocytes with NIH3T3 cells, and ES cells with STO cells.
Materials and Methods
Preparation of MCLs
The magnetite (Fe3O4; average particle size, 10 nm) used as the core of the MCLs was donated by Toda Kogyo (Hiroshima, Japan). Magnetic characterizations of the magnetite at 796 kA/m (at room temperature) were 2.0 kA/m, 63.9 Am2/kg, and 2.6 Am2/kg for coercivity, saturation flux density, and remanent flux density, respectively. The MCLs were prepared from colloidal magnetite and a lipid mixture consisting of N-(α-trimethylammonioacetyl)-didodecyl-D-glutamate chloride (TMAG, a cationic lipid), dilauroylphosphatidyl-choline (DLPC), and dioleoylphosphatidyl-ethanolamine (DOPE) in a 1:2:2 molar ratio, as described previously. 26 The average particle size of MCLs was 150 nm by dynamic light scattering. 29
MCL uptake by cells
Mouse NIH3T3 cells (American Tissue Culture Collection, Manassas, VA) and STO cells (Riken Cell Bank, Tsukuba, Japan) were cultured using Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 0.2 mg/mL streptomycin sulfate, and 100 U/mL potassium penicillin G. HUVECs were purchased from Kurabo (Osaka, Japan) and cultured in Humedia-EG2 (Kurabo). The cells were cultured at 37°C in a humidified atmosphere of CO2 and 95% air. Uptake of MCLs by the cells was measured as described previously. 27 Briefly, the cells (NIH3T3, 2 × 106 cells; STO, 5 × 106 cells; or HUVECs, 1 × 106 cells) were first seeded into 100-mm cell culture dishes (Greiner Bio-One, Frickenhausen, Germany). After 24 h incubation, the medium was replaced with a fresh one containing MCLs with predetermined net magnetite concentrations (0–200 pg/cell), and the cells were further incubated. The cell samples were collected at 4 h (NIH3T3 cells), 24 h (HUVECs), or periodically (1, 4, and 24 h, STO cells) after MCL addition, to measure iron concentration and cell number using potassium thiocyanate 30 and trypan blue dye exclusion, respectively.
Magnetic capture of cells labeled with MCLs
For magnetic labeling of cells, MCLs (net magnetite concentration, 100 pg/cell) were added to NIH3T3 cells, STO cells, or HUVECs, and the cells were incubated for 4 h (NIH3T3 cells) or 24 h (STO cells and HUVECs). For magnetic capture of cells, the cells were suspended in 1 mL of culture medium in 1.5-mL sterile polypropylene tubes, and magnetic force was applied to the tube using a cylindrical neodymium magnet with surface magnetic induction of 1200 or 4000 G (diameter, 50 mm; height, 10 mm). Magnetic flux density measured at the position of the cell pellet in the tube using a gauss meter (F.W. Bell, Orlando, FL) was 924 and 2660 G, for the 1200 and the 4000 G magnet, respectively. After 3 min, the supernatant was aspirated, and the cell pellet was resuspended in 1 mL of the medium, and the cell number was counted by trypan blue dye exclusion. The percentage of the cell capture rate was determined by the following equation:
Effect of culture period on magnetic capture of NIH3T3 cells
The effect of the culture period on magnetic cell capture was measured using NIH3T3 cells treated with or without mitomycin C (Nacalai Tesque, Kyoto, Japan). In the case without mitomycin C treatment, NIH3T3 cells (2 × 106 cells) were seeded into 100-mm cell culture dishes (Greiner Bio-One). When they were approximately 80% confluent, the medium was replaced with a fresh one containing MCLs (100pg/cell), and the cells were incubated for 4 h. Then, the cells were replated with 6 × 105 cells in 10 mL of fresh medium in 100-mm cell culture dishes (Greiner Bio-One), and a serial subculturing of the cells was performed every 2 days. The cell samples were collected periodically, and cell number, iron concentration, and magnetic cell capture rate were measured as described above.
For the experiments with mitomycin C treatment, NIH3T3 cells (1 × 106 cells) were seeded into 60-mm cell culture dishes (Greiner Bio-One). When they were approximately 80% confluent, the medium was replaced with a fresh one containing MCLs (100 pg/cell), and the cells were incubated for 4 h. After the cells were treated with 10 μg/mL mitomycin C for 3 h, the medium was replaced with fresh medium without mitomycin C. Then, the medium was changed every 2 days. The cell samples were collected periodically, and cell number, iron concentration, and magnetic cell capture were measured as described above.
The microscopic observation of magnetite was performed by Prussian blue staining. 31 Briefly, the cells were washed twice with phosphate-buffered saline (PBS), and fixed in 10% formaldehyde solution. Cells were stained with a 5% potassium ferrocyanide solution for 5 min at room temperature. Culture dishes were then filled with a chlorhydric ferrocyanide solution for 20 min at room temperature. The cells containing iron were stained in blue.
Layered coculture of hepatocytes with various cells
Primary rat hepatocytes were obtained from Wistar rats by a two-step collagenase perfusion method. This experiment was reviewed by the Ethics Committee on Animal Experiments of the Faculty of Engineering, Kyushu University. Details of the hepatocyte culture medium and protocols were reported previously.
6
For the magnetic labeling of cells, MCLs (100 pg/cell) were added to NIH3T3 cells, STO cells, or HUVECs, and the cells were incubated for 4 h (NIH3T3 cells) or 24 h (STO cells and HUVECs). Hepatocytes and the MCL-labeled cells were prestained with 5-chloromethylfluoroscein diacetate (CMFDA, green fluorescent probe; Molecular Probes, Eugene, OR) and 5-(and-6)-(((4-chloromethyl)benzoyl)amino)tetramethylrhodamine (CMTMR, red fluorescent probe; Molecular Probes), respectively. A layered coculture of hepatocytes with the MCL-labeled cells was performed first by seeding freshly isolated hepatocytes onto wells of 35-mm collagen-coated tissue culture dishes (Asahi Techno Glass, Tokyo, Japan) at 5 × 105 cells/dish, followed by seeding 5 × 105 of MCL-labeled cells onto the monolayer of hepatocytes. One day after the beginning of coculture, the cells were harvested, and magnetic cell separation was performed. After trypsin treatment, the cells were suspended in 1 mL of medium in 1.5-mL sterile polypropylene tubes, and magnetic force was applied to the tube for 3 min using a 4000 G magnet. The number of hepatocytes and other cells in the supernatant was counted using a hematocytometer under a fluorescence microscope (Olympus, Tokyo, Japan). Purity and recovery yield of target cells (hepatocytes in this case) were determined by the following equations:
Coculture of keratinocytes with NIH3T3 cells
A spontaneously immortalized human keratinocyte cell line, HaCaT 32 , was cultured at 37°C under a humidified atmosphere of 5% CO2 and 95% air in DMEM supplemented with 5% FBS, 0.1 mg/mL streptomycin sulfate, and 100 U/mL potassium penicillin G. For magnetic labeling of NIH3T3 cells, MCLs (100 pg/cell) were added to NIH3T3 cells, and the cells were incubated for 4 h. Then, the cells were treated with 10 μg/mL mitomycin C for 3 h. NIH3T3 and HaCaT cells were prestained with CMFDA (green fluorescent probe; Molecular Probes) and CMTMR (red fluorescent probe; Molecular Probes), respectively. HaCaT cells were cocultured with NIH3T3 cells by seeding the cells (HaCaT, 2 × 105 cells; NIH3T3, 2 × 105 cells) into 35-mm tissue culture dishes (Greiner Bio-One). One day after the beginning of coculture, the cells were harvested and magnetic separation of the cells was performed. The cells were suspended in 1 mL of medium in 1.5-mL sterile polypropylene tubes, and magnetic force was applied to the tube using a 4000 G magnet. After 3 min, the number of HaCaT and NIH3T3 cells in the supernatant was counted using a hematocytometer under a fluorescence microscope (Olympus), and purity and recovery yield of HaCaT cells were determined.
Coculture of ES cells with STO cells
Murine ES cells (H1 strain 33 obtained from the Riken Cell Bank) were maintained at 37°C under a humidified atmosphere of 5% CO2 and 95% air in DMEM supplemented with 15% KnockOut™ serum replacement (Gibco, Grand Island, NY), 4 mM L-glutamine, 0.1 mM nonessential amino acids (Gibco), 0.1 mM 2-mercaptoethanol, 0.05 mg/mL streptomycin sulfate, and 0.05 mM potassium penicillin G (ES medium). The undifferentiated state was maintained by adding 1000 U/mL recombinant murine LIF (ESGRO; Chemicon, Temecula, CA). To prepare the magnetically labeled STO feeder layer, MCLs (100 pg/cell) were added to the STO cells when they were at approximately 80% confluency, and the cells were incubated for 24 h. After the cells were treated with 10 μg/mL mitomycin C for 3 h, the cells were replated at 1 × 106 cells/dish into gelatin-coated 35-mm tissue culture dishes. Coculture was performed by seeding ES cells (5 × 105) onto the STO feeder cells. After 2 days from the beginning of the coculture, the cells were harvested, and magnetic separation of the cells was performed. The cells were suspended in 1 mL of medium in 1.5-mL sterile polypropylene tubes, and magnetic force was applied to the tube using a 4000 G magnet. After 3 min, the number of ES and STO cells in the supernatant was counted using a hematocytometer under a fluorescence microscope (Olympus), and the purity and recovery yield of the ES cells were determined.
For alkaline phosphatase (ALP) staining of ES cells, the cells in coculture were washed twice with PBS, fixed in 0.25% glutaraldehyde solution for 5 min at room temperature, and exposed to a solution containing naphthol AS-MX phosphate (Sigma, St. Louis, MO) as a substrate and Fast Violet B Salt (Sigma) as a coupler for 20 min at 37°C under a humidified atmosphere of 5% CO2 and 95% air. Cells showing ALP activity were stained dark violet.
Embryoid bodies (EBs) were induced using the hanging-drop method.34,35 After the magnetic separation, the cells were resuspended in the ES medium without LIF at a concentration of 7000 cells/mL. A total of 15 μL drops of the cell suspension (48 drops were prepared per experiment) were placed on the lid of a bacterial grade 100-mm plastic dish (As One, Osaka, Japan). The lid was turned upside down and placed on the bottom part, which was filled with PBS and then incubated at 37°C under a humidified atmosphere of 5% CO2 and 95% air. After 2 days, the cells were transferred into 24-well culture plates (Greiner Bio-One) containing 0.5 mL ES medium without LIF. In this study, a single and spherical cell aggregate was called EB, and the EB formation efficiency was defined as follows:
For fluorescence microscopy of EBs, STO cells were prestained with CMFDA (green fluorescent probe; Molecular Probes).
Statistical analysis
The Mann–Whitney rank sum test was used to evaluate the statistical significance of differences. Statistically significant differences were established at a p-value below 0.05.
Results
MCL uptake and magnetic capture of cells
For magnetic labeling of cells, MCLs were added to the medium in NIH3T3, STO, and HUVEC cultures. In our previous reports, the uptake of MCLs for NIH3T3 cells 28 and HUVECs 36 reached a maximum at 4 and 24 h after MCL addition, respectively, and the amounts were 18 and 32 pg of magnetite per cell, respectively. In the present study, we measured the uptake of MCLs in STO culture, when MCLs were added to the medium at 100 pg/cell. As shown in Figure 1A, the cells took up MCLs rapidly, and the uptake reached a maximum at 24 h after MCL addition (29 pg of magnetite/cell). Thus, in subsequent magnetic labeling experiments, the cells were cultured in the presence of MCLs for 4 h (NIH3T3 cells) and 24 h (STO cells and HUVECs), respectively.
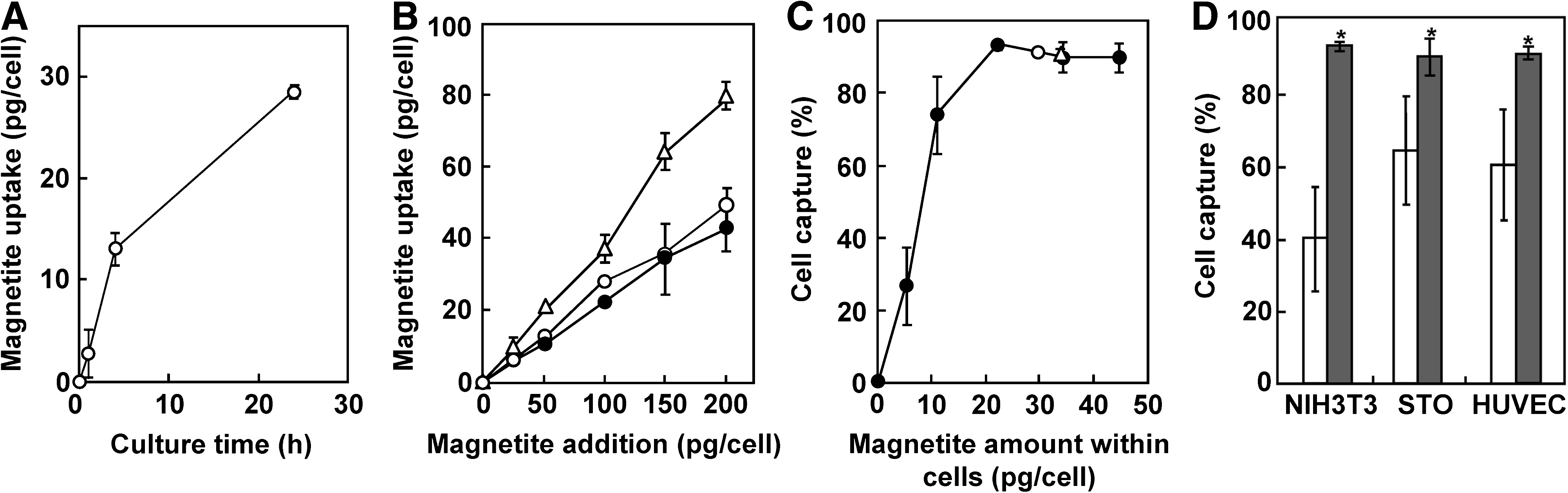
Magnetite uptake and magnetic cell capture. (
When MCLs at concentrations of 0–200 pg/cell were added in NIH3T3, STO, and HUVEC cultures, the uptake of MCLs linearly increased (Fig. 1B). Using the cells labeled with various amounts of MCLs, the cell capture efficiency was measured using a 4000 G magnet. As shown in Figure 1C, the capture efficiency of NIH3T3 cells increased with the amount of magnetite incorporated into the cells, and reached 93% when the magnetite amount in the cells was 21 pg/cell (magnetite addition, 100 pg/cell; Fig. 1B). When the magnetite amounts were more than 21 pg/cell, the cell capture efficiency of NIH3T3 cells was greater than 90%, and this relationship was also observed for STO cells (cell capture, 90%; magnetite amount within cells, 29 pg/cell; magnetite addition, 100 pg/cell) and HUVECs (cell capture, 91%; magnetite amount within cells, 34 pg/cell; magnetite addition, 100 pg/cell). The cell capture efficiency also depended on the magnetic field intensity at the position of the cell pellet in the tube. As shown in Figure 1D, significantly higher efficiencies of cell capture were achieved using a 4000 G magnet (surface magnetic induction, 4000 G; magnetic flux density at the cells, 2660 G) rather than using a 1200 G magnet (surface magnetic induction, 1200 G; magnetic flux density at the cells, 924 G). Taken together, when MCLs were added to the cells at a level of 100 pg/cell, greater than 90% of the cell capture could be achieved using a 4000 G magnet. In subsequent experiments, for the magnetic labeling of cells, MCLs were added at 100 pg/cell in NIH3T3, STO, and HUVEC cultures.
Effect of culture period on magnetic capture of NIH3T3 cells
In some cocultures, a long-term cultivation may be required to enhance or maintain cellular functions. Therefore, the effect of the culture period on the magnetic capture efficiency of cells was examined using NIH3T3 cells. As shown in Figure 2A, the cells labeled with MCLs (100 pg/cell) proliferated exponentially throughout the 7-day culture period, which was comparable to the proliferation of cells without MCLs (data not shown). On the other hand, the magnetite amount within the cells exponentially decreased and became undetectable within the 7-day culture period (Fig. 2B). Prussian blue staining of the cells revealed that the amount of magnetite within the cells was diluted on day 1 and had almost disappeared by day 5 (Fig. 2C). In these experiments, the average doubling time of the cells (td, 0.93 day; calculated from Fig. 2A) and the half-life period of the magnetite amount within the cells (t1/2, 0.92 day; calculated from Fig. 2B) were almost equivalent, suggesting that the dilution of the magnetite particles within the cells was caused by cell proliferation. Thus, the percentage of cell capture decreased throughout the culture period, and by day 4, most of cells could hardly be captured (Fig. 2D).
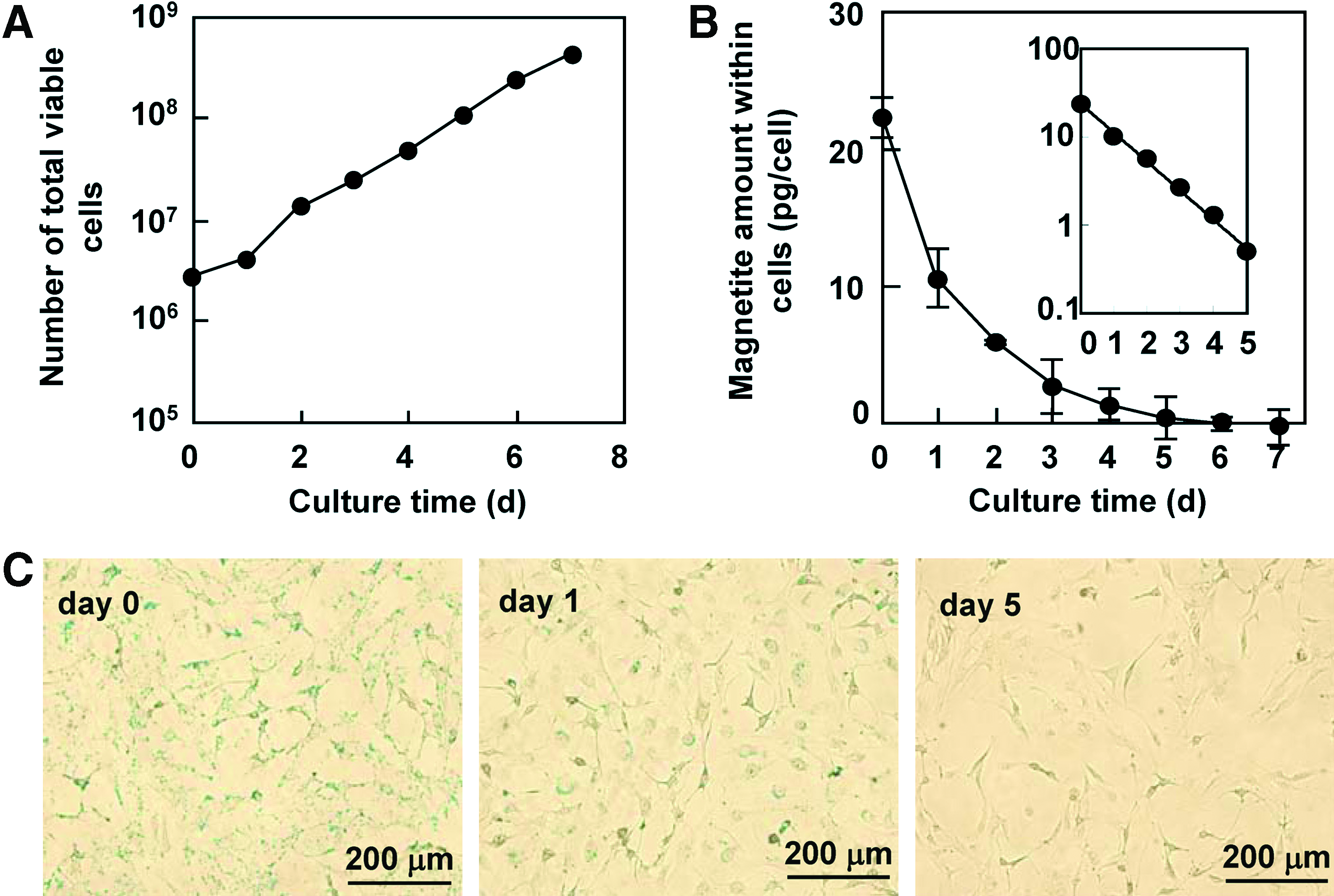
Effect of culture period on magnetic capture of NIH3T3 cells. (
For the experiments of cocultures using feeder cells, the cells acting as the feeder were often treated with mitomycin C to cease their cell proliferation. Next, we examined the effect of the culture period on the magnetic capture efficiency of NIH3T3 cells treated with mitomycin C. As shown in Figure 3A and B, the cells treated with mitomycin C did not proliferate, and the magnetite amount within the cells was maintained at around 20 pg/cell throughout the 7-day culture period. No obvious dilution of the amount of magnetite particles within the cells was observed by microscopic detection using Prussian blue staining of the cells (Fig. 3C). These results suggest that the magnetite particles within the cells mainly decreased due to cell proliferation and were maintained for at least for 7 days if the cells were incapable of cell division. The cells labeled with MCLs could be captured at greater than 90% efficiency using a 4000 G magnet even on day 7 (Fig. 3D).
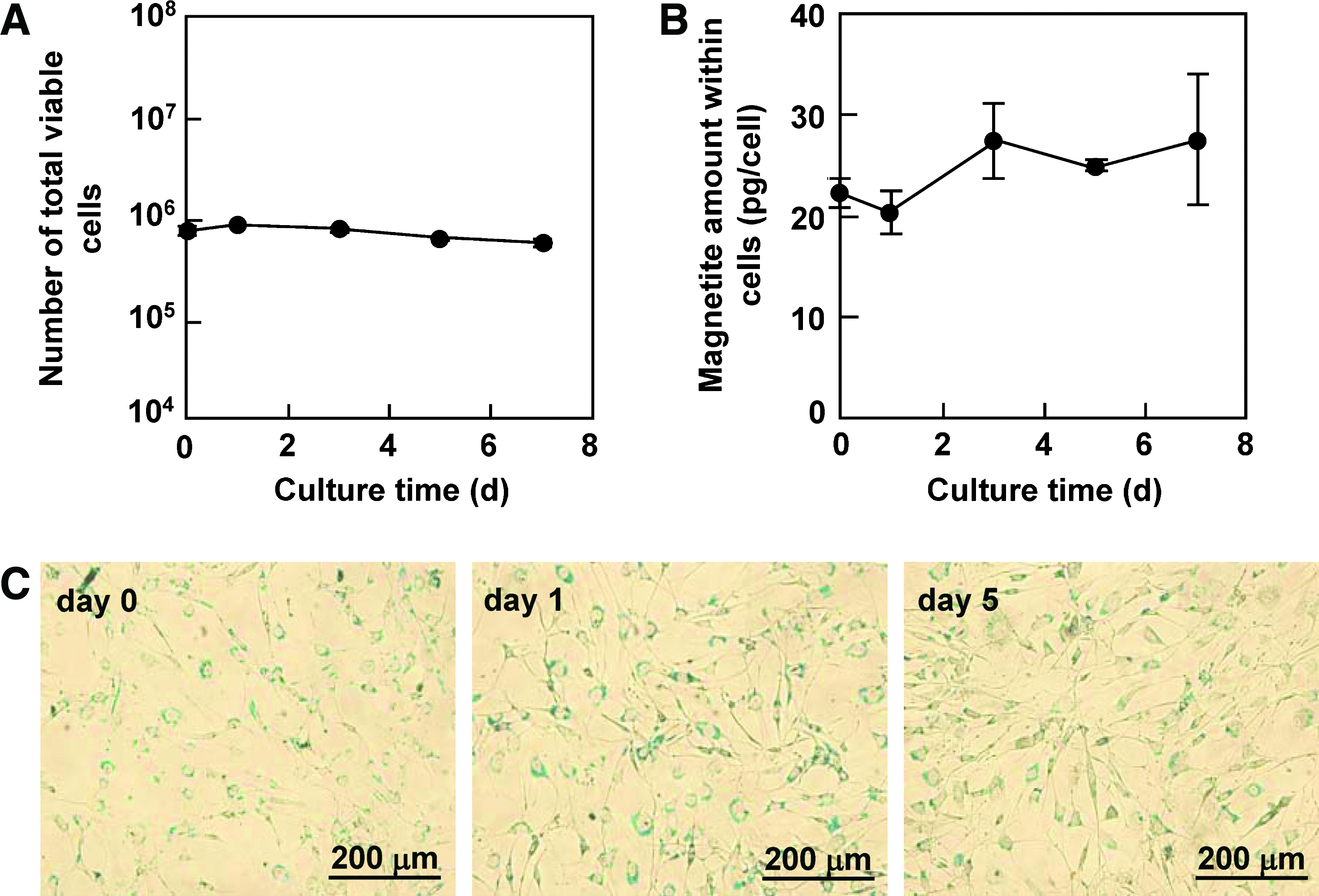
Effect of culture period on magnetic capture of mitomycin C–treated NIH3T3 cells. (
Layered coculture of hepatocytes with various cells
Layered coculture of hepatocytes with various cells such as NIH3T3 37 or endothelial cells 9 has been reported to be an effective method to enhance liver-specific functions of hepatocytes. In the present study, cells (NIH3T3 cells, STO cells, or HUVECs) labeled with MCLs were inoculated on a monolayer of rat primary hepatocytes. As shown in Figure 4A–D, NIH3T3 cells adhered onto a monolayer of hepatocytes, resulting in a layered coculture of hepatocytes with NIH3T3 cells. After 24 h from the beginning of coculture, the proportion of hepatocytes before magnetic separation was around 50% (45% for NIH3T3 cells, 58% for STO cells, and 49% for HUVECs; Fig. 4E), suggesting that no obvious proliferation of NIH3T3 cells, STO cells, or HUVECs was observed, because the coculture was started with a 1:1 ratio of these cells to hepatocytes (which are known not to divide). Hepatocytes were separated from the cocultures with approximately 90% purities (Fig. 4E and Table 1) and greater than 91% recovery yields (Table 1) using a 4000 G magnet.

Layered coculture of hepatocytes with various cells. NIH3T3 cells, STO cells, or HUVECs labeled with MCLs were cultured on a monolayer of rat primary hepatocytes. Bright-field (
Coculture of keratinocytes with NIH3T3 cells
HaCaT keratinocytes were cocultured with mitomycin C–treated NIH3T3 cells as a model system, and we investigated whether recovery of HaCaT cells could be achieved by magnetic separation of NIH3T3 cells labeled with MCLs. Equal numbers (2.0 × 105 cells) of HaCaT and NIH3T3 cells were seeded in the dish and cultured for 1 day. As shown in Figure 5A–D, epithelial islands formed by HaCaT cells were surrounded by extended NIH3T3 cells. As shown in Figure 5E, no obvious proliferation of NIH3T3 cells was observed, while HaCaT cells proliferated at 1 day after the beginning of coculture. The proportion of HaCaT cells in the harvested cell suspension before magnetic separation was 73% (Fig. 5F), which corresponded to the percentage calculated from the number of cells in Figure 5E. In the coculture system, HaCaT cells were magnetically separated with 97% purity (Fig. 5F and Table 1) and a 99% recovery yield (Table 1).
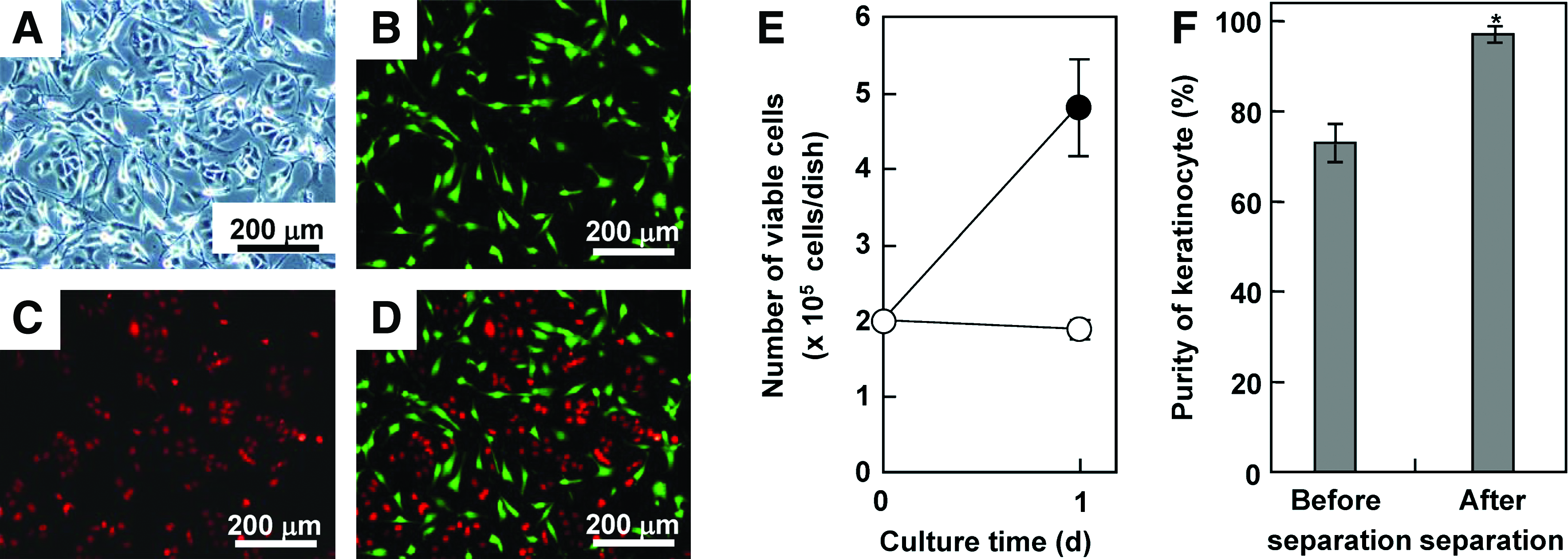
Coculture of keratinocytes with NIH3T3 cells. Equal numbers (2.0 × 105 cells) of HaCaT and NIH3T3 cells were seeded in the dish and cultured for 1 day. Bright-field (
Coculture of ES cells with STO cells
Mouse ES cells (5 × 105 cells/dish) were cultured onto the MCL-labeled STO feeder layer (1 × 106 cells/dish) for 2 days. ALP staining revealed that typical colonies of undifferentiated ES cells were cultured on the STO feeder layer (Fig. 6A). Due to the rapid growth of ES cells, the proportion of ES cells in the harvested cell suspension before magnetic separation was 71.6% (Fig. 6B). By magnetically separating the STO cells, the ES cells were separated with 96% purity (Fig. 6B and Table 1) and approximately 100% recovery yield (Table 1).
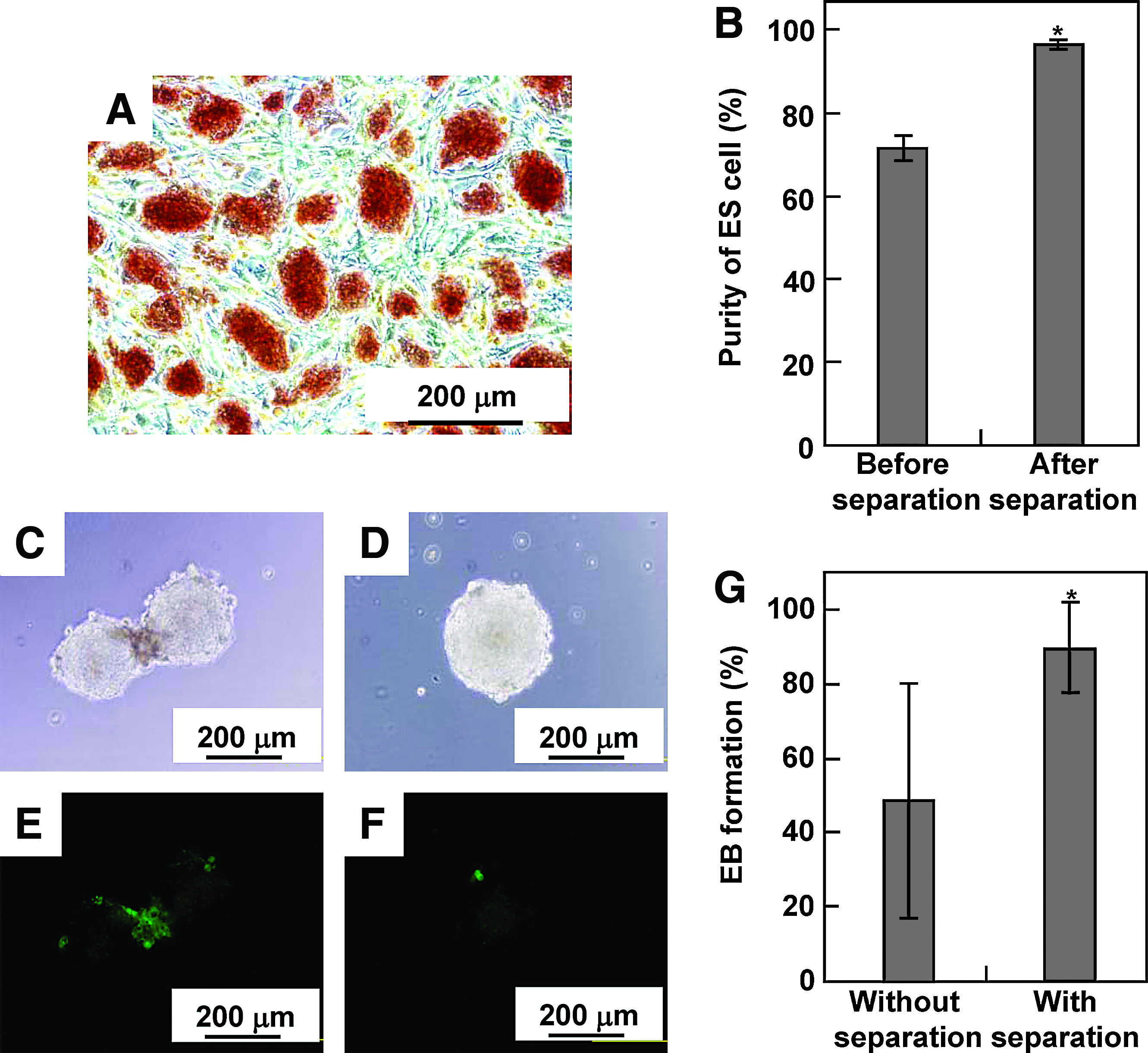
Coculture of ES cells with STO cells. Mouse ES cells (5 × 105 cells/dish) were cultured onto a mitomycin C–treated STO feeder layer (1 × 106 cells/dish) for 2 days. (
It is often necessary to separate and recover target cells for further analysis and application. When ES cells are transferred to suspension culture without feeder cells and LIF, ES cells begin to differentiate and form 3D multicellular aggregates called EBs. Because the EB plays an important role in the differentiation process of ES cells, we investigated whether the magnetic separation of ES cells from feeder cells had any effects on EB formation. A single EB, whose average diameter was 197.1 ± 6.5 μm, was formed in a droplet by the hanging-drop method (Fig. 6D). As shown in Figure 6G, the formation efficiency of EB without magnetic separation was 50%, which was comparable with a previous report by Kurosawa et al. 38 On the other hand, the formation efficiency of EB after magnetic separation was 90% (Fig. 6G), which was significantly higher than that without the magnetic separation. Fluorescence microscopy revealed that some STO cells were observed at the periphery of cell aggregates without magnetic separation (Fig. 6C, E) and appeared to hamper the EB formation, suggesting that the failure of EB formation was due to the presence of STO cells within ES cell aggregates.
Discussion
There are two major strategies for separation of a specific cell type: positive isolation of the cells of interest and negative isolation where unwanted cells are removed. In the present study, we chose negative isolation because the intact target cells may be preferable to further analysis. For the magnetic labeling of cells, we used magnetite nanoparticles coated with cationic liposomes consisting of TMAG, DLPC, and DOPE, which are known to have low immunogenicity and high stability, are inexpensive to manufacture, and currently have been used as a gene delivery system of plasmids for clinical applications of interferon-β gene therapy for malignant glioma in Japan. 39 However, because the cationic liposomes used in this study potentially have a toxic effect on cells at high concentrations, 40 negative isolation might be preferable. The magnetic cell separation technique using MCLs enables label-free, simple, and rapid recovery of target cells through magnetic removal of other cells, which has advantages over existing cell separation methods such as FACS and immunomagnetic cell sorting systems. We demonstrated the advantages of the negative isolation method by using authorized representative coculture systems where growth of nontarget cells is inhibited artificially or by close contacts with target cells, but this point may also show a limitation in wide range of necessities of cell separations from cocultured cells where nontarget cells grow.
Interestingly, in the long-term culture of NIH3T3 cells labeled with MCLs, it was revealed that an average doubling time of cells (td, 0.93 day) and half-life period of magnetite amount within the cells (t1/2, 0.92 day) were almost equivalent (Fig. 2). On the other hand, the cells treated with mitomycin C maintained the magnetite amount within the cells throughout the 7-day culture period (Fig. 3). These results indicated that the dilution of magnetite particles within the cells was not due to exocytosis, but dominantly caused by cell proliferation. Cationic polymers including liposomes are known to be taken up by cells via endocytosis. Huth et al. 41 reported that polyethyleneimine–magnetite conjugates were taken up by cells via clathrin-dependent and caveolae-mediated endocytosis. However, there are few reports on how these conjugates are excreted from the cells. Characterization of the cellular uptake and intracellular trafficking of MCLs still remains to be elucidated.
There are two types of operational modes of magnetic cell separation systems: column-based and tube-based operations. The column-based operation system usually utilizes smaller, nanosized particles and therefore requires that cells are passed through a magnetized iron-mesh column to increase cell-capture capacity. The tube-based operation system utilizes larger, micron-sized beads to enhance the magnetic attraction force to a magnet. On the other hand, although the magnetite particles used in the present study were very small at 10 nm in diameter, the tube-based operation was utilized and achieved higher cell-capture efficiency (Fig. 1C). The high cell-capture capacity of MCLs was due to the encapsulation of the small magnetite nanoparticles into the larger cationic liposomes and the accumulation of a large amount of the MCLs into target cells by electrostatic interactions. As shown in Figure 1B, the uptake amount of MCLs differed among cell types. In the previous studies, tumor cell lines showed a higher uptake of MCLs than cultured primary cells. For example, when MCLs were added to the cells at 100 pg/cell, the uptake amounts of magnetite by tumor cells were as follows: T-9 rat glioma cells, 55 pg/cell 26 ; U251 human glioma cells, 60 pg/cell 42 ; mouse renal cell carcinoma, 40 pg/cell. 43 In this study, two fibroblast cell lines (NIH3T3 and STO cells) and primary HUVECs showed relatively low uptake amounts of MCLs (NIH3T3 cells, 22 pg/cell; STO cells, 25 pg/cell; HUVECs, 38 pg/cell; Fig. 1B). Some tumor cells possess high endocytotic activity,44,45 suggesting that the uptake of MCLs depends on the endocytotic activity of the target cells. Nevertheless, MCL is a superior tool for magnetic labeling of nonspecific cells. Thus, this is a great advantage in applying the technique because surface antigens and a specific antibody are not always available for each cell type.
In normal liver tissues in vivo, each hepatocyte has intimate interactions, not only with adjacent hepatocytes, but also with nonparenchymal cells in a 3D manner. However, it is difficult to reconstruct such 3D interactions in vitro. To overcome this difficulty, Okano and coworkers developed a culture and recovery method of cell sheets on a thermoresponsive culture surface using poly(N-isopropylacrylamide), and coculture of double-layered cell sheets was achieved by overlaying an endothelial cell sheet onto a monolayer of hepatocytes on the culture surface deposited with extracellular matrices. 9 Thus, this kind of cell sheet engineering may be adaptable to tissue engineering. Alternatively, we applied a magnetic force to construct a heterotypic, layered coculture system of rat hepatocytes and HAECs. 10 HAECs labeled with MCLs specifically accumulated onto a monolayer of hepatocytes at sites where a magnet was positioned, and then adhered to form a heterotypic, layered construct with tight and close cell-to-cell contact. This cocultured construct significantly enhanced albumin secretion by hepatocytes compared with that in homotypic cultures of hepatocytes. Similarly, Nishikawa et al. reported that enhanced liver functions, such as albumin secretion, ammonia removal, and urea synthesis, were observed when NIH3T3 cells were cultured on a monolayer of rat hepatocytes. 37 Thus, the layered coculture of hepatocytes with another type of cell is a simple and effective method, and this kind of coculture may be useful for basic biological research of cell-to-cell communication. In the present study, NIH3T3 cells, STO cells, or HUVECs labeled with MCLs were cultured on a monolayer of rat hepatocytes. As shown in Figure 4E, no obvious proliferation of the cells was observed, which was consistent with the previous result using HAECs. 10 In the present study, samples containing around 90% pure hepatocytes were achieved by magnetic separation. Further analysis of hepatocytes separated from the coculture should be the next subject of research. Gene expression analysis of cells separated from the coculture may reveal signaling pathways of heterotypic cell-to-cell interactions.
Feeder cells are often necessary for successful cultivation of keratinocytes and ES cells. The essence of the method is to use mitomycin C–treated cells as the feeder, to keep their viability without allowing cell proliferation. Because the cells treated with mitomycin C maintained magnetite amounts within the cells (Fig. 3), feeder cells labeled with MCLs were successfully removed from coculture by magnetic separation with high purity of the target cells (Table 1). In general, ES cells are enriched by plating to remove contaminating feeder cells, which is based on the differential adhesion properties of ES cells and feeder cells on uncoated dishes. More than one cycle of plating and replating is necessary to reduce the contamination level. Meanwhile, this process causes a reduction in recovery of ES cells during each replating cycle. On the other hand, the single magnetic separation achieved approximately 100% recovery with high purity of ES cells (Table 1), suggesting that the magnetic separation using MCLs is a rapid and precise method. The residual feeder cells seem to secrete antidifferentiation factors and exert their influence in the local microenvironment. Thus, a high-purity ES cell population is beneficial to any subsequent experimental manipulations. As an example of a further application of the separated target cells, we attempted to form EBs using ES cells separated from the coculture of STO feeder cells labeled with MCLs. Significantly higher EB formation efficiency was observed by magnetic separation of feeder cells, although the precise mechanism of why ES cells containing feeder cells were hampered in EB formation is not known. EB formation involves cell adhesion and cellular remodeling, which implicates the involvement of cell adhesion molecules. In particular, E-cadherin, a homophilic cell-to-cell adhesion molecule, seemed to be involved in this process. E-cadherin−/− ES cells have been reported to be unable to grow and form cell aggregates. 46 Because E-cadherin is a homophilic adhesion molecule, cells expressing different cadherin molecules such as N- and P-cadherins tend to distinguish each other when artificially mixed in vitro. 47 Because STO cells do not express E-cadherin, STO cells remained within EB aggregates may interfere with E-cadherin–mediated EB formation.
In the present study, 94% purity and 98% recovery yield of target cells on average were successfully achieved, as shown in Table 1. In some applications, particularly in the medical field, higher purity of up to 100% would be required. Recent reports suggested that the use of a xenosupport system introduced considerable disadvantages with respect to exploiting the therapeutic potential. 48 To achieve higher purity and recovery yield, a magnet with a higher magnetic induction is required, as shown in Figure 1D. Because magnetic induction is inversely proportional to the distance, it is important to set the magnet close to the cells. In the present study, we chose the tube-based system, and the magnetic flux density at the position of the cell pellet in the tube was 2660 G, which was much less than the surface magnetic induction (4000 G). Thus, an alternative cell isolation device with higher magnetic induction should be used to obtain higher purity and recovery yield, especially for clinical applications.
In conclusion, we proposed a new technique for the magnetic separation of target cells in coculture systems, in which MCLs were used for magnetic labeling of nontarget cells, and high purity and recovery yield of target cells were achieved. These results indicate that the magnetic cell separation technique using MCLs is applicable for coculture systems in the tissue engineering field.
Footnotes
Acknowledgments
The authors would like to thank Toda Kogyo Co. for supplying the magnetite. This work was supported in part by Grants-in-Aid for Scientific Research (nos. 19686049 and 20034043) from the Japan Society for the Promotion of Science (JSPS).
Disclosure Statement
No competing financial interests exist.