
Abstract
Bioluminescent quantification of viable cells inside three-dimensional porous scaffolds was performed in vitro and in vivo. The assay quantified the bioluminescence of murine stem (C3H10T1/2) cells tagged with the luciferase gene reporter and distributed inside scaffolds of either soft, translucent, AN69 polymeric hydrogel or hard, opaque, coral ceramic materials. Quantitative evaluation of bioluminescence emitted from tagged cells adhering to these scaffolds was performed in situ using either cell lysates and a luminometer or intact cells and a bioluminescence imaging system. Despite attenuation of the signal when compared to cells alone, the bioluminescence correlated with the number of cells (up to 1.5 × 105) present on each material scaffold tested, both in vitro and noninvasively in vivo (subcutaneous implants in the mouse model). The noninvasive bioluminescence measurement technique proved to be comparable to the cell-destructive bioluminescence measurement technique. Monitoring the kinetics of luciferase expression via bioluminescence enabled real-time assessment of cell survival and proliferation on the scaffolds tested over prolonged (up to 59 days) periods of time. This novel, sensitive, easy, fast-to-implement, quantitative bioluminescence assay has great, though untapped, potential for screening and determining noninvasively the presence of viable cells on biomaterial constructs in the tissue engineering and tissue regeneration fields.
Introduction
The distribution of viable and functioning cells throughout the biomaterial scaffolds is important for the in vivo success of cell/biomaterial constructs, but it has remained a challenge for tissue engineering applications. Methods currently used to assess the number and distribution of viable cells within 3D scaffolds include DNA assays, cell viability stains, and assays that determine aspects of cell metabolism, for instance, lactate dehydrogenase or adenosine triphosphate (ATP).3–7 These methods can be used for cells cultured on scaffolds in vitro or cell constructs retrieved after implantation into animal models. However, despite their efficacy, these methods are sample destructive and do not discriminate between implanted cells and invasive host cells; in addition, the presence of the scaffold material may interfere with the assays used. For these reasons, there is a need for standardized testing methods to quantify in situ and monitor cell viability within scaffolds. Development and availability of noninvasive approaches for the evaluation of the cell content and distribution throughout cell-scaffold constructs is needed for basic research and, most importantly, clinical applications.
To this end, gene-reporter technologies (involving, e.g., the labeling of cells with a bioluminescent reporter gene) are promising because they enable the noninvasive detection of cells on scaffolds and, thus, provide crucial information on cell-containing constructs pertinent to tissue engineering and tissue regeneration applications. 8 Luciferase, the reporter protein produced by genetically modified cells, is easily assayed using ultrasensitive light detectors. Because of its high sensitivity, bioluminescence technology has recently proven its usefulness in tracking stem cells on material scaffolds transplanted in live animals for tissue engineering purposes.8–10 However, no information regarding correlations between the bioluminescence and the number of cells on the respective material scaffolds was reported in these pioneering studies. The presence of scaffold material may impair cell bioluminescence because of photon absorption by the scaffold material; moreover, there may be limited bioavailability of the D-luciferin substrate, a critical aspect for successful luminescence, for cells inside scaffolds that have small pores.
The present study addressed these questions by evaluating the efficacy and reliability of bioluminescence in quantitatively assaying the number of viable cells inside 3D synthetic material scaffolds in vitro and in vivo. Mesenchymal stem cells (MSCs) were tagged with the firefly luciferase reporter gene under the control of a constitutively active promoter. We quantified and characterized luminescence under both cell-destructive (cell lysates using a reader luminometer) and nondestructive (intact cells using an imaging device) conditions after seeding cells onto two different porous scaffold materials, specifically, a polymeric translucent soft hydrogel and an opaque hard ceramic. The specific aims of the present study were to establish, under both in vitro and in vivo conditions, (i) a correlation between bioluminescence and the number of cells within scaffolds and (ii) a long-term (3 and 8 weeks for in vitro and in vivo studies, respectively) method of monitoring cell proliferation. For the sake of comparison, the number of cells inside 3D scaffolds in vitro was quantitatively measured by fluorescence emitted by enhanced green fluorescent protein (eGFP) reporter gene contained in the cells used the present study.
Materials and Methods
Retroviral vectors, cell culture, and viral transfection
The retroviral vectors rMLV-LTR-eGFP and rMLV-LTR-Luciferase (109 viral particles/mL; Genethon, Evry, France) used for cell labeling contained the eGFP and luciferase gene sequences, respectively. The C3H10T1/2 murine embryonic (MSCs; American Type Culture Collection) were cultured in Eagle's basal medium (BME) supplemented with 10% fetal calf serum (FCS), penicillin (100 U/mL), and streptomycin sulfate (100 μg/mL) (all from PAA Laboratories, Les Mureaux, France).
The C3H10T1/2 cells were genetically modified in two successive gene transfer steps using rMLV-LTR-eGFP and then rMLV-LTR-Luciferase. First, the cells were plated at a density of 8 × 103 cells/cm2 and cultured under standard cell culture conditions in BME (containing 10% FCS and antibiotics) in a humidified, 37°C, 5% CO2/95% air environment for 24 h. The cells were then rinsed twice with phosphate buffered saline (PBS), infected with BME containing 20% FCS, 400 ng/mL polybrene (Sigma-Aldrich, Saint Quentin Fallavier, France), and 2 × 106 viral particles/cm2 (250 particles/cell), and cultured again under standard conditions for 24 h. After a medium change, the cells were expanded according to established protocols.11,12 An aliquot of these cells was then transfected with the second vector using the aforementioned protocol. The stably transfected C3H10T1/2 cells were further cloned using the limited dilution method. One clonal cell line (C3H10-LF33), which exhibited the highest fluorescence and luciferase expression, was selected for the present study.
AN69 hydrogel and coral scaffolds
The two porous scaffold materials used in the present study, specifically a polymeric translucent soft hydrogel and an opaque hard ceramic, were chosen because of their very different physicochemical characteristics. Disk-shaped AN69 hydrogels (diameter 6 mm; height 2 mm) were donated by Dr. Jiri Honiger (Saint-Antoine Hospital, Paris, France). The AN-69 polymer is an acrylonitrile and sodium methallyl sulfonate copolymer. The hydrogels were prepared by phase-inversion of a polymer solution containing 6% AN-69 polymer, 91% dimethylsulfoxide, and 3% physiological saline (0.9% NaCl) solution (w/w/w). 13 The AN69 hydrogel scaffolds had pores with a mean diameter of 500–1000 μm and a porosity of 77%. The hydrogels were sterilized via emersion in 10% Dialox® in physiological saline (v/v) and thoroughly rinsed using sterile physiological saline.
Cube-shaped (3 × 3 × 3 mm3) scaffolds of natural Porites coral (Biocoral®) were donated by Inoteb, Inc. (Levallois-Perret, France). The scaffolds consisted of 99% calcium carbonate in the form of aragonite and had open, communicating pores with a mean diameter of 100–300 μm, and a porosity of 49%.14,15 Before cell seeding, these scaffolds were sterilized by autoclaving.
Scaffold seeding
Small numbers (up to 150,000) of C3H10-LF33 cells were used to seed the scaffolds; this cell number was chosen from a range tested because it proved to be the maximum number of cells that would adhere on the substrates of interest to the present study in preliminary experiments. Unless specified, each material scaffold was seeded with 100,000 cells. For seeding, each scaffold was immersed in an aliquot (50 μL) of cells in BME (containing 10% FCS, antibiotics, and 25 mM of Acide 4-(2 hydroxyethyl)-1-piperazine ethane sulfonique (HEPES)). The constructs were maintained under slight vacuum at room temperature for 15 min, and then under standard cell culture conditions for 4 h. At that time, 1 mL culture medium was added per construct. After 8 h of incubation, unless otherwise specified, the scaffolds were noninvasively imaged, washed three times with 1 mL of PBS (to eliminate the D-luciferin), and stored at −20°C until the in vitro luciferase and fluorescence assays were performed. For comparison purposes, a series of the same number of C3H10-LF33 cells in suspension in 50 μL PBS were prepared in parallel and treated in the same way as scaffolds seeded with cells.
In vitro cell proliferation
One day after seeding 100,000 cells onto each scaffold, the constructs were placed in a custom-made, rotational, oxygen-permeable bioreactor system that enabled cell culture under dynamic conditions while providing sufficient oxygen tension. 16 A thin coating of silicone elastomer (Sylgard 184, Dow Corning) was applied to the inside of a 50 mL polypropylene centrifuge tube. After curing, a 30 × 40 mm2 window was cut into the cylindrical surface of the tube to allow gas exchange. Twenty-one scaffolds were placed in each bioreactor. Three bioreactors were prepared per scaffold material: one contained unloaded scaffolds (negative control), and the other two contained the C3H10-LF33 cell constructs. These gas-permeable bioreactors were placed on a roller apparatus (rotating speed 6 rpm) in a tissue culture incubator and were maintained under standard cell culture conditions for the duration of the specific experiment. After 3, 7, 10, 14, and 21 days of culture, three scaffolds from each bioreactor were removed, frozen, and maintained at −20°C until the luciferase and fluorescence assays were performed.
In vivo implantation
The European Guidelines for Care and Use of Laboratory Animals (EEC Directives 86/609/CEE of 24.11.1986) were observed during all aspects of the animal studies. One day after cell seeding, two constructs were aseptically implanted in the backs of 8-week-old nude mice (Janvier, Legenest-Saint-Isle, France), which had been anaesthetized with ketamine (100 mg/kg; Ketalar®; Panpharma, Virbach, France) and xylazine (10 mg/kg; Rompun® 2%; Bayer, Lille, France). The backs were cleaned with ovidone–iodine (Betadine®; Vetoquinol, Lure, France), and the scaffolds inserted into pouches created between the muscles and the aponeurotic layer. The soft tissues at the implantation sites were closed with interrupted resorbable sutures.
At the end of the study, the animals were euthanized using CO2. For comparisons of the noninvasive and invasive measurements, explants were harvested after 1, 2, and 3 weeks of implantation and stored at −20°C until the in vitro luciferase and fluorescence assays were performed. For monitoring the in vivo cell proliferation on the implanted scaffolds, the animals were imaged (as described in the Nondestructive bioluminescence imaging section) twice a week for 8 weeks before they were sacrificed. Each subcutaneous implantation site was opened postmortem, and integration of each scaffold in the surrounding tissues was analyzed.
In vitro luciferase assay
Quantitative determination of in vitro luminescence from C3H10-LF33 cells was performed using cell lysates (destructive cell conditions). Cells, either in situ on porous scaffolds or in suspension, were lysed using 200 μL lysis buffer (containing 0.1% Triton, 25 mM Gly-Gly, 15 mM MgSO4, 4 mM EGTA [pH 7.4], and 1 mM dithiothreitol). After the addition of 200 μL D-luciferin substrate (200 μM; Interchim) in 25 mM Gly-Gly (pH 7.8), 200 μM acetyl CoA, 200 μM adenosine triphosphate (ATP), 3 mM MgSO4, and 2 mM dithiothreitol, the luminescence of each sample was measured using a luminometer (Berthold Lumat 9501). Luminescence data were expressed in relative light units over a 20-s integration period.
Fluorescence assay
Fluorescence was measured on aliquots of the same samples used for the bioluminescence analysis. After the in vitro luciferase assay, each sample was subjected to three freeze/thaw cycles before the fluorescence (relative fluorescence units (RFU)) from 200 μL of supernatant was determined using a FL600 fluorescent plate reader (Bio-tek Instruments, Colmar, France) at excitation and emission wavelengths of 485 nm and 510 nm, respectively.
Nondestructive bioluminescence imaging
Bioluminescence imaging (BLI) was carried out using an IVIS 100 imaging system (Xenogen, Alameda, CA). For in vitro imaging, either cell suspensions or seeded scaffolds were loaded in individual wells of 96-well black plates, immersed with 200 μL of D-luciferin solution (300 μg/mL in PBS), and then immediately imaged using the medium binning setting of the instrument for 10 min. There was no time lapse between addition of the D-Luciferin substrate and sample imaging because previous time-course experiments determined that the maximum bioluminescence signal occurred within the first 2 min. For in vivo imaging, an aliquot (50 μL) of D-luciferin (30 mg/mL in PBS) was injected subcutaneously in the area surrounding each implant in animals that has been anesthetized with isoflurane. Ten minutes after the D-luciferin substrate injection, the animals in the prone position were imaged using the medium binning setting for a 10 min exposure for each implant. The acquisition time was reduced down to 1 min when the obtained images were over-saturated. To assess the putative correlation between the number of cells on implanted scaffolds and light emission, the scaffolds were imaged as follows: in vitro before implantation; in vivo the day of implantation (day 0); and 2 days postimplantation. To compare noninvasive and invasive measurements, animals were imaged after 1, 2, and 3 weeks of implantation before sacrifice. To monitor cell proliferation on the implanted scaffolds in vivo, the animals were imaged on the day of implantation (day 0) as well as twice a week for an 8-week period postimplantation. Light emission was quantified using Living Image software (version 2.50.2; Xenogen). Each frame depicted the bioluminescent signal in pseudocolor superimposed on the respective gray-scale photographic image. Data from a region of interest surrounding each implant were manually selected on the frames and were reported as the total photon flux defined by the net photon count emitted per second per centimeter squared per steradian (p/s/cm2/sr).
Statistical analyses
Each in vitro experiment was performed twice in triplicate. Numerical results were reported as mean ± standard error of the mean. The in vivo experiments were analyzed comparing pre- and postimplantation (n = 5), in vivo noninvasive and in vitro cell-destructive (n = 5), and the time-course of cell proliferation results (n = 6). Linear regression analysis was performed to obtain correlation coefficients. Quantitative data were analyzed by two-way analysis of variance. The confidence interval was set at 95% and the significance level at p < 0.05. Two methods of cell quantification were compared by analysis of the differences between the two measurement systems using Bland–Altman plots. 17 For these purposes, and because the raw data of the signals from each scaffold tested using different methods (specifically, IVIS BLI, luminometer, or fluorescence) were in different units, each set of data was normalized using their respective averages. The reported data were, therefore, dimensionless and could be compared by the Bland–Altman analysis. Statistical analyses were conducted using the Statgraphics centurion version XV.2 (Statpoint, Inc., Herdon, VA).
Results
Bioluminescence and fluorescence correlate with the number of cells in suspension
Luciferase expression in C3H10-LF33 cells proved to be a function of cell number. The correlation between the emitted signal and cell number (in the range of 10 to 106 cells) was linear (R2 = 0.996 and R2 = 0.985 for luminometer and IVIS BLI measurements, respectively; data not shown). Expression of eGFP was also determined to be linearly correlated (R2 = 0.991) with cell numbers (in the range of 103 to 106 cells; data not shown). Bioluminescence, detected with either a luminometer or IVIS BLI, required the presence of a minimum of 30 LF33 cells, whereas fluorescence required a minimum of 500 LF33 cells.
Bioluminescence and fluorescence correlates with the number of cells adhering to material scaffolds in vitro
Nonadhering cells accounted for less than 5% of the number of seeded cells on each scaffold tested (data not shown). The bioluminescence of the lysates from adhering cells exhibited a linear relationship with the number of seeded cells. Compared to the light emitted from the same number of cells in suspension, however, an attenuation was observed (Fig. 1A, B). Specifically, this attenuation (determined from the ratio of the slope values of plotted standards) was 73 ± 5% and 65 ± 3% for cells adhering to AN69 hydrogels and coral scaffolds, respectively. Fluorescence measurements of lysates from samples subjected to cellular bioluminescence measurements provided similar results: the slope of standard plots for cells alone was higher than the slope of standard plots for the seeded scaffolds (Fig. 1C, D). Specifically, an attenuation of 31 ± 4% and 47 ± 5% was observed for cells adhering to AN69 hydrogels and coral scaffolds, respectively. The correlations between the IVIS BLI and luminometer signals from C3H10-LF33 cells in suspension (R2 = 0.972), adhering to AN69 hydrogels (R2 = 0.933), and adhering to coral (R2 = 0.843; Fig. 2A) were good (p < 0.001). The correlations between the bioluminescent (from luminometry) and fluorescent signals from C3H10-LF33 cells in suspension (R2 = 0.841), adhering to AN69 hydrogels (R2 = 0.947), and adhering to coral (R2 = 0.884; Fig. 2C) were also good (p < 0.001). The three methods tested in the present study to quantify the cell number were compared using Bland–Altman plots, which examined the agreement between IVIS BLI and luminometer signals (Fig. 2B) as well as between luminometer and fluorescent signals (Fig. 2D); the results depict the difference between the two methods versus the average signal from two methods for each individual sample tested. The Bland–Altman analysis confirmed that the data obtained by the two procedures were in good agreement. These results provide strong support for the use of in vitro bioluminescence (using either a BLI system or a luminometer) and fluorescence to quantify cells cultured on various biomaterial scaffolds.
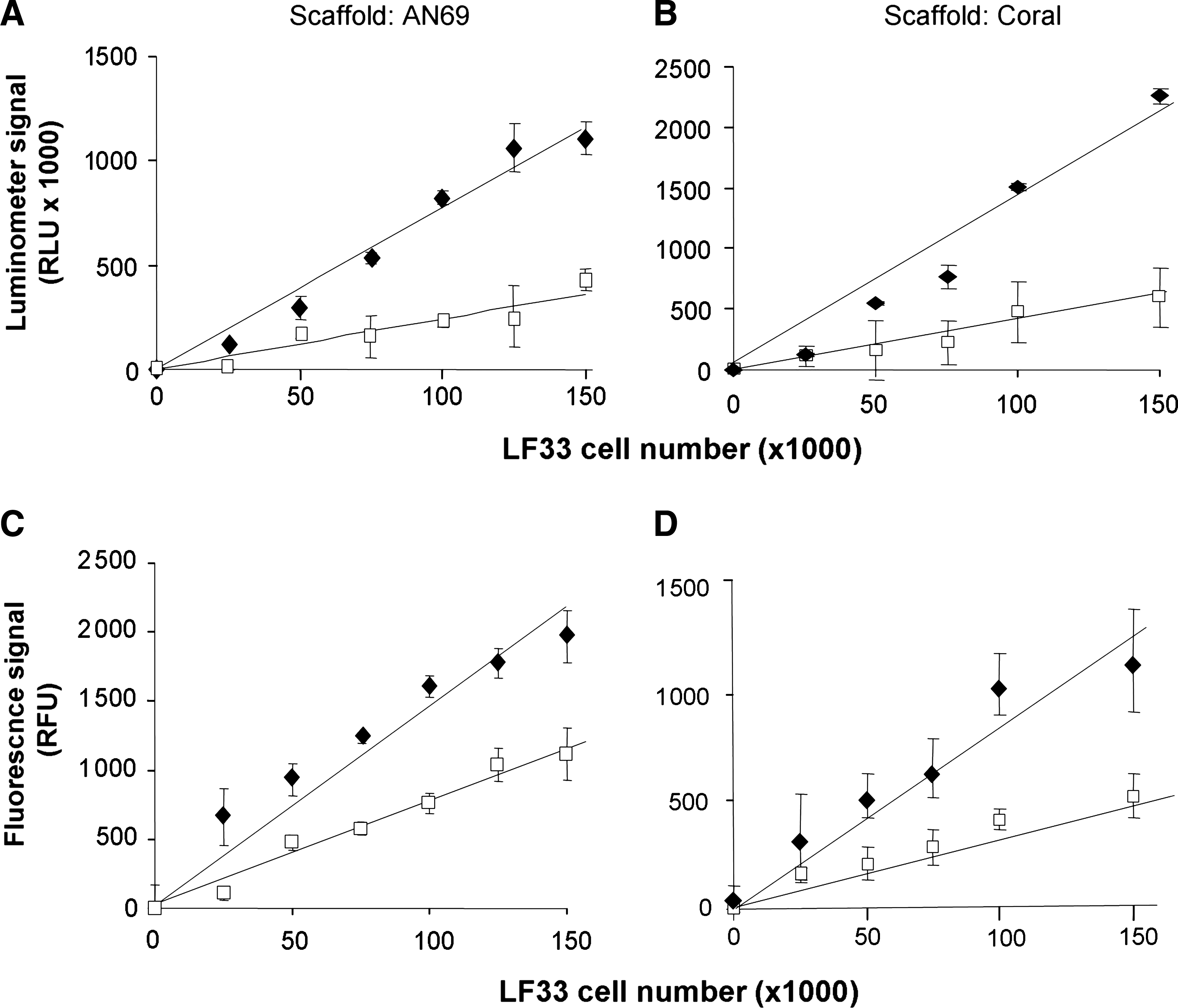
In vitro luminescence and fluorescence from C3H10-LF33 cells adhering onto scaffolds. Measurements from a luminometer (
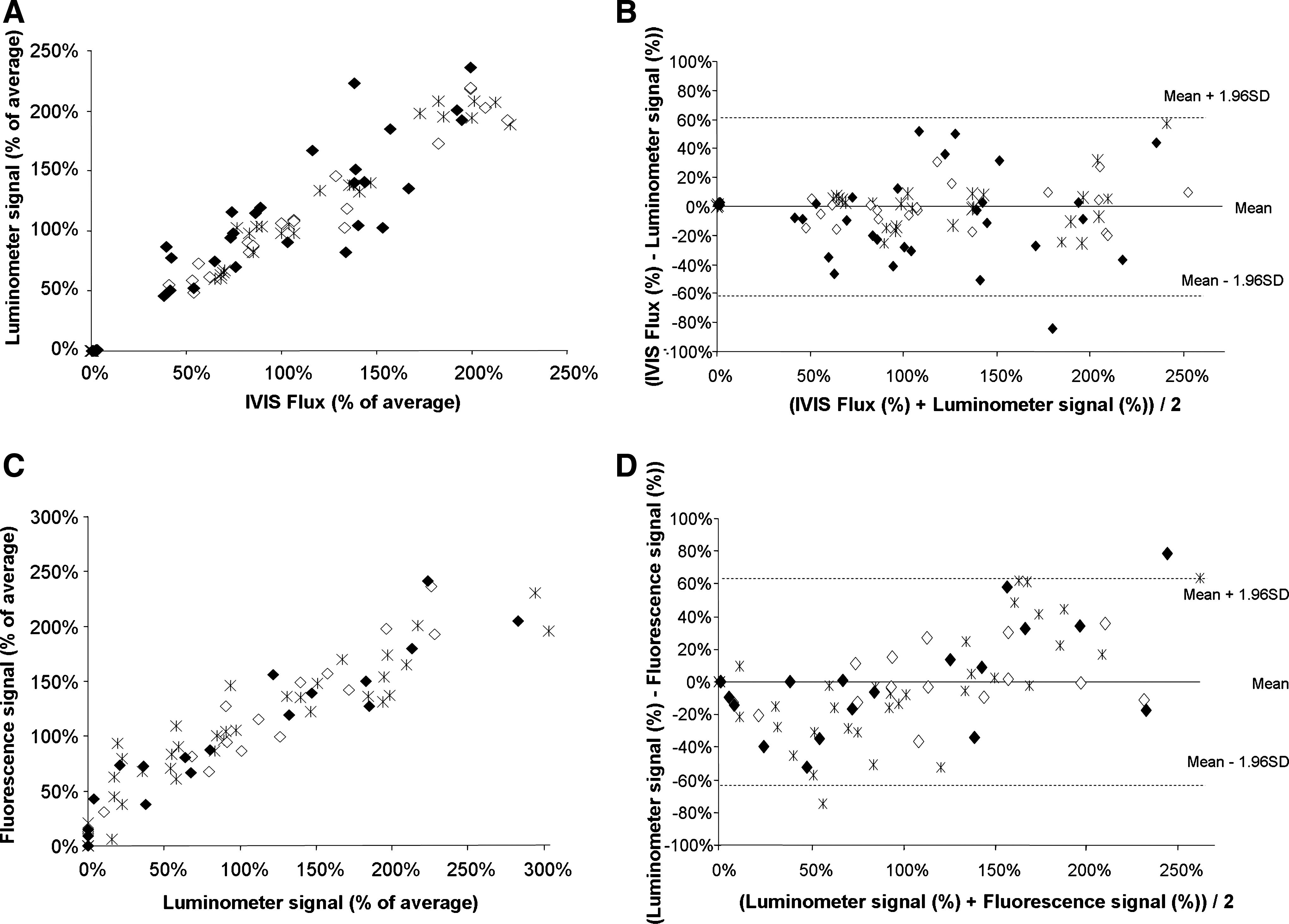
Comparisons between the in vitro IVIS BLI and luminometer (in lysates) signals (
Luminometry enables monitoring of in vitro cell proliferation on scaffolds
Quantitative cell proliferation data were obtained for each time point during the 21-day in vitro culture by measuring the light emitted from the lysates of cells cultured on the scaffolds tested and subtracting the signal background obtained from respective scaffolds without cells. The results provide evidence of enhanced cell proliferation on AN69 hydrogel compared to coral scaffolds (Fig. 3A). A sustained increase in the quantitative bioluminescence of cells cultured on AN69 hydrogels was observed; specifically, an 18-fold increase in bioluminescence was observed at day 21 compared to day 0. In contrast, the bioluminescence of cells cultured on coral scaffolds yielded only a fourfold increase during the same time period. Similar cell proliferation trends as those shown in Figure 3A were obtained for the fluorescence measurements of cell lysates over the 21-day culture (Fig. 3B).
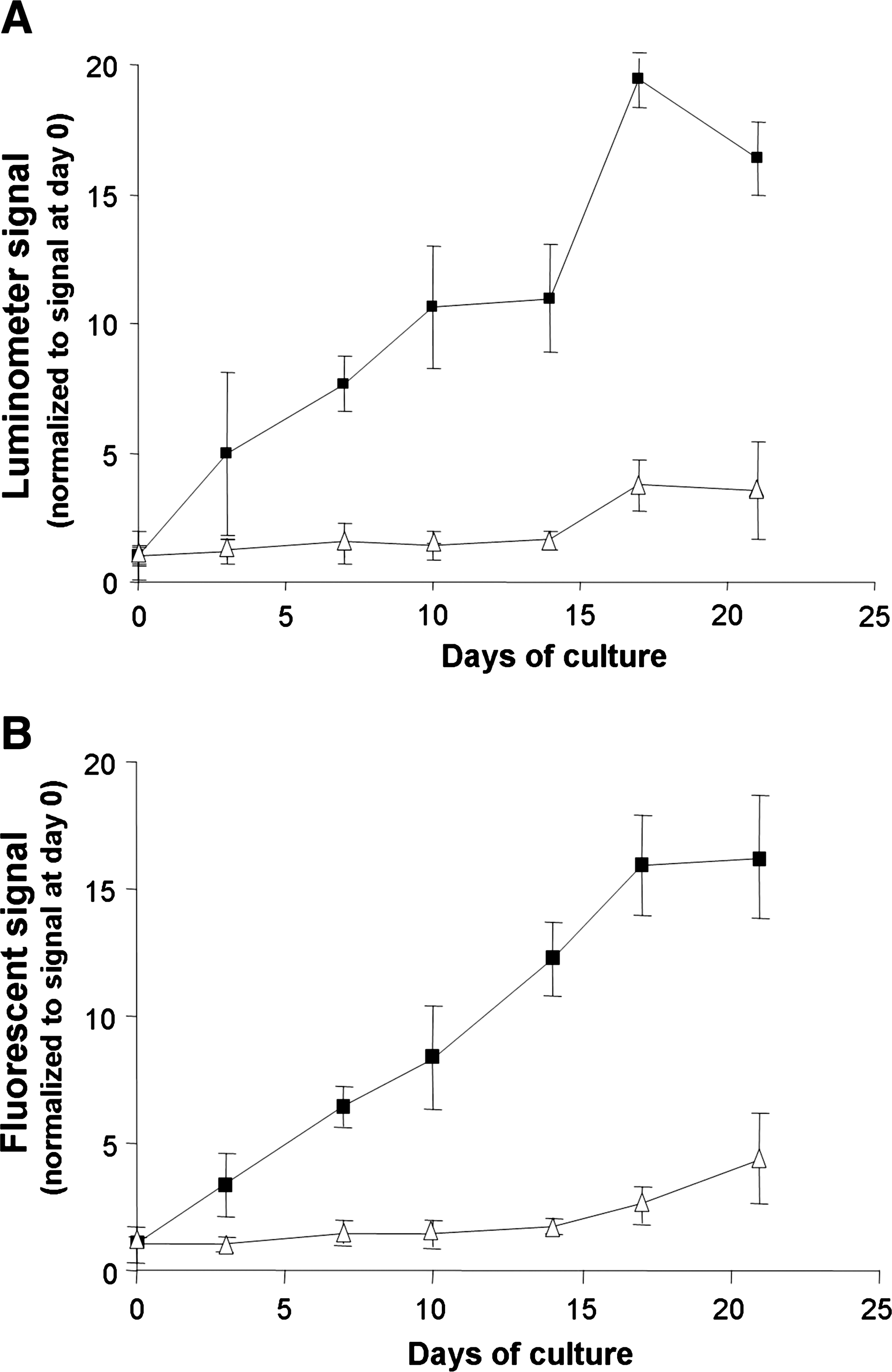
Time course of the in vitro proliferation of C3H10-LF33 cells seeded onto either AN69 hydrogels (black squares) or coral scaffolds (white triangles) in a rotational bioreactor (6 rpm). At each time point, the luminescence (
IVIS BLI signals correlate with the number of cells adhering to material scaffold in vivo
To determine the optimal BLI signal, a time course of light emission using IVIS BLI was determined in mice implanted with cell-containing scaffolds, locally injected with luciferin, and sequentially imaged for an acquisition time of 5 min, at 5-min intervals for a total of 20 min. During the first 5 min, a strong bioluminescent signal was observed, increased during the next 5 min, and reached a plateau at the interval 10–15 min postadministration of luciferin (data not shown). For this reason, in subsequent studies with implanted scaffolds, in vivo IVIS BLI data were acquired within 10 min of luciferin administration.
To establish that the IVIS bioluminescence is correlated with cell number in vivo, cells adhering to either AN69 hydrogels or coral scaffolds were subcutaneously implanted in nude mice. For all scaffolds seeded with cells, the IVIS signal was higher than background, which was measured using the same type of scaffold without cells. An analysis of IVIS images of scaffolds loaded with LF33 cells, both in vitro (Fig. 4A, C) and in vivo (Fig. 4B, D), provided evidence of linear relationships between the level of detected photons fluxes and the number of cells present on each construct (Fig. 4E, F). A linear quenching effect was observed for the IVIS signal transmitted from implanted scaffolds compared to the respective in vitro signals obtained before implantation of the same scaffolds. The range of the observed attenuation was different for the two types of scaffolds. Specifically, a twofold (p < 0.005) and threefold (p < 0.005) attenuation was observed for cells cultured on AN69 and coral, respectively. An additional fourfold decrease for cells seeded on coral occurred between days 0 and 2 (p < 0.005) (Fig. 4E, F). Comparison of the preimplantation and in vivo IVIS BLI signals for each construct demonstrated a moderately strong linear correlation for cells seeded on either AN69 (R2 = 0.740, p < 0.001; Fig. 4G) or coral (R2 = 0.532, p < 0.005; Fig. 4H).
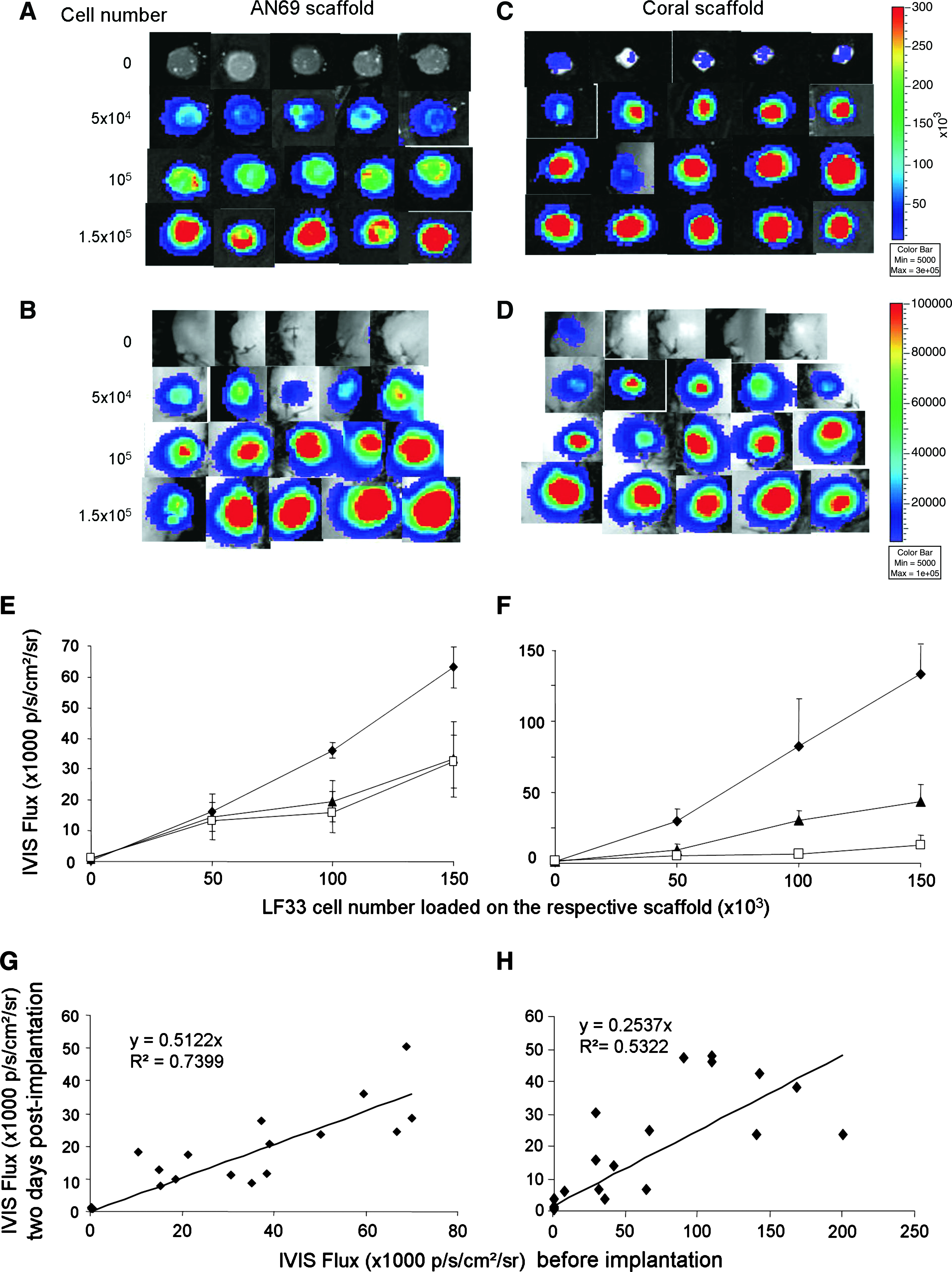
In vitro and in vivo bioluminescence detection in C3H10-LF33 cells on scaffolds using the IVIS BLI system. Representative bioluminescent images of 5 × 104 to 1.5 × 105 C3H10-LF33 cells adhering to either AN69 hydrogels (
To establish that the in vivo noninvasive bioluminescence signal is comparable with the in vitro cell destructive (lysates) measurements, the in vivo IVIS BLI signals from cell-seeded scaffolds after 1, 2, and 3 weeks of implantation were compared to the in vitro luciferase activities from cell lysates measured from the same scaffolds after the sacrifice of the animals. These results also evidenced a moderately strong linear correlation (specifically, R2 = 0.587 for AN69 scaffolds and R2 = 0.873 for coral scaffolds; p < 0.001; Fig. 5A). The Bland–Altman plot (Fig. 5B) was used to determine the agreement between the bioluminescence signals measured by both the noninvasive and cell destructive (lysates) procedures used in the present study. With the exception of 3 out of 40, all measurements were observed within the 95% confidence interval of the mean values of the observed differences between the two methods tested. Taken together, these results suggest that IVIS bioluminescence provides a reliable noninvasive measurement of cells on scaffolds.
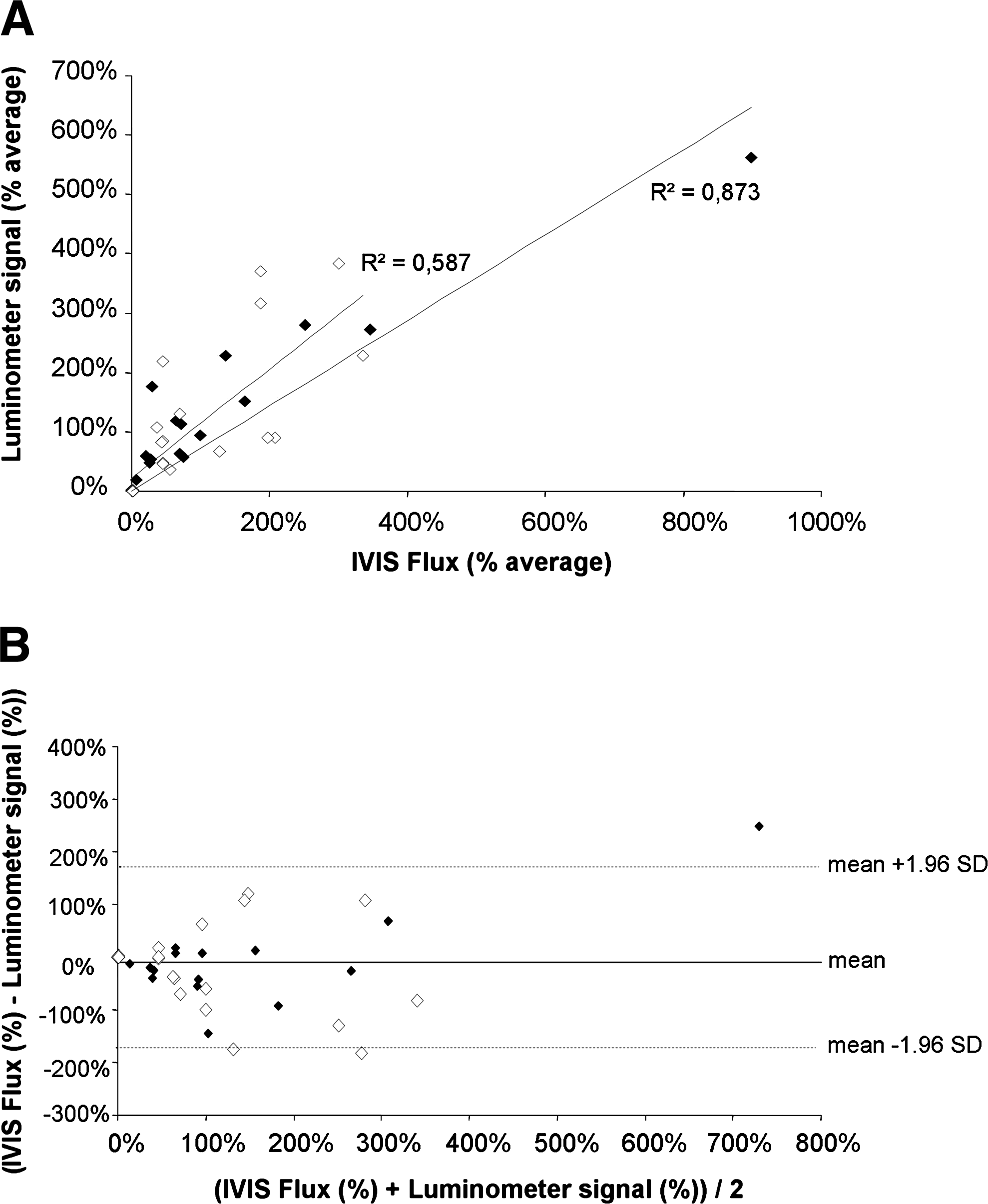
Comparison between in vivo noninvasive bioluminescence signals (using the IVIS BLI system) and in vitro cell destructive (lysates) bioluminescence signals (using a luminometer). The in vivo IVIS BLI signals from cells seeded onto either AN69 hydrogels (white diamonds) or coral scaffolds (black diamonds) after 1, 2, and 3 weeks of implantation (n = 5 for each time point and material tested) were measured before the sacrifice of the animals and excision of the constructs. After lysis of cells from each explant, the in vitro luminometer signal was measured. A linear regression model (
IVIS BLI enables the noninvasive monitoring of in vivo cell proliferation on scaffolds
To noninvasively determine in vivo cell proliferation, the constructs of interest of the present study were subcutaneously implanted in mice. Representative IVIS images from each test group are shown in Figure 6A and B. The photon flux from empty scaffolds was subtracted from respective measurement of the same constructs with cells. The net photon count emitted per second per centimeter squared per steradian (p/s/cm2/sr) from each seeded scaffold was plotted versus time postimplantation (Fig. 6C, D). In addition, to account for variations in the number of cells seeded, the photon fluxes were normalized to those obtained on day 0 (Fig. 6E, F). The IVIS BLI images and corresponding data demonstrating cell viability and proliferation within the scaffolds were monitored over an 8-week period postimplantation. During the first 3–6 days after implantation, the IVIS bioluminescence of cell-loaded AN69 and coral scaffolds decreased by approximately 70% and 80%, respectively, but increased after 1 week postimplantation (Fig. 6C–F). Despite variations in the signals from each scaffold over the 8-week period postimplantation, overall cell proliferation was different on the two scaffold materials. After the cell number decrease observed during the first week postimplantation, cells seeded on AN69 scaffolds survived but proliferated at a slow rate over the duration of the study; the average final (at day 59) cell population was 2.5-fold higher than the initial seeding number. In contrast, cells seeded on coral scaffolds exhibited a high rate of proliferation: after the first 3-day decrease, the initial cell number (as detected by the IVIS BLI signal) was reached between days 6 and 18; subsequently, the cell number increased, yielding on average a 12-fold increase at day 59 (p < 0.005 compared to cell-seeded AN69 implants at the same time point).
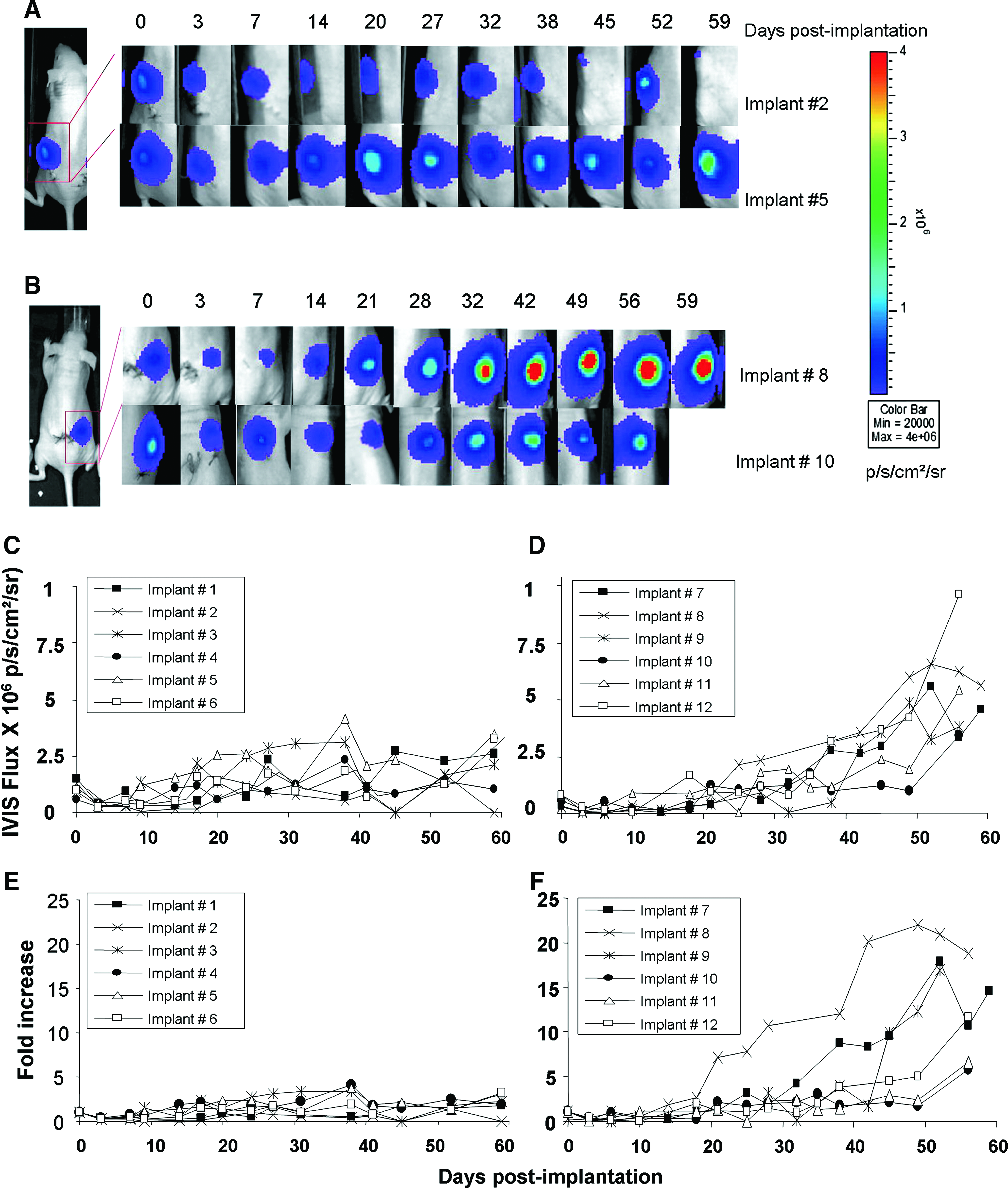
Time course of the in vivo proliferation of C3H10-LF33 cells on scaffolds during 59 days of implantation. These are representative BLI images of cell proliferation in subcutaneously implanted AN69 hydrogels (
Discussion
In the present study we assessed the efficacy of bioluminescence technology to quantify in situ the number of viable stem cells (MSCs) distributed inside 3D porous material scaffolds both in vitro and in vivo. The two porous scaffold materials examined, specifically a polymeric translucent soft hydrogel and an opaque hard ceramic, were chosen because of their very different physicochemical characteristics that are critical for the luminescent signal transmission from these cell-seeded constructs. In addition, these two scaffold materials exhibited excellent properties of cell adherence and proliferation in published studies.14,18–22
The correlation between the bioluminescent signal from cell lysates and the number of cells adhering onto each of the two scaffold materials tested was linear (Fig. 1). Similarly, the correlation between the IVIS bioluminescent signal and the theorical number of cells seeded onto scaffolds under nondestructive conditions was also linear both in vitro and in vivo (a noninvasive determination in mice model) (Fig. 4). Compared to cells in suspension, the total signal emitted by cells adhering to the scaffolds tested was reduced by two-thirds (Fig. 1). Such a decrease is likely due, in part, to the absorption of photons by the scaffold material. Moreover, other aspects may affect the extent of the observed signal attenuation caused by the presence of the scaffold material, such as the physicochemical features of the material, the size, shape, and porosity of the scaffold.
In the present study, the in vitro bioluminescence of cells adhering to AN69, a translucent material, was 1.8-fold lower than that of cells adhering to coral, an opaque material, proving that parameters other than the apparent opacity of the material influence the bioluminescence of adhered cells. Specifically, variations in the cell distribution profiles on 3D scaffolds are likely very important since light emitted will be scattered and, therefore, attenuated to a greater or lesser extent, depending on the location of the origin of the signal inside the scaffold. Histological analysis revealed that the MSCs were distributed throughout the AN69 hydrogels including the center of these scaffolds after 6 weeks of culture; in contrast, on coral scaffolds the cells were present close to the outer surfaces (periphery) of the construct (K. Oudina; personal communication). The observed bioluminescence results may reflect these differences in cell distribution. Additionally, incomplete cell lysis of the cells dispersed inside the scaffolds tested and/or impaired access of substrate reagent to luciferase are other limitations that might have influenced the bioluminescence signal measurement. Compared to cell constructs imaged in vitro, the in vivo IVIS bioluminescence of implanted cell constructs was decreased by two to threefold; this additional attenuation was caused by the presence and interference of overlying living tissues in the subcutaneous location in mice.
The nondestructive in vitro IVIS bioluminescence of cells on scaffolds correlated with the respective noninvasive in vivo IVIS signals (Fig. 4), providing evidence for the reliability of the luminescence measurement in living animals. Additionally, comparisons between (either in vitro [Fig. 2A, B] or in vivo [Fig. 5]) nondestructive bioluminescence signals and those obtained from in vitro cell lysates measurements were performed by linear regression and Bland–Altman analyses, and demonstrated that the two measurement techniques tested are comparable.
Current practice for in vitro assessment of cell number within 3D porous matrices is a difficult, if not impossible, task relying on DNA measurement, a method that, to the best of our knowledge, has not yet been validated for measurements of cell number in engineered tissues. DNA quantification methodology does not require genetic labeling of cells, but is sample destructive and has a limited linear dynamic range (usually 50 to 500,000 cells maximum). 23 Moreover, because the dye used stains all nucleic acids, that is, both DNA and RNA, accurate DNA quantification requires additional treatment of samples with RNase, an expensive and time-consuming process. 24 Lastly, the presence of extracellular matrix and/or a scaffold material further complicates proper and complete DNA extraction.
The number of eGFP-labeled C3H10T1/2 cells was also quantified in the present study by measuring the emitted fluorescence; this is a straight-forward method widely used in the field of tissue engineering. In contrast to bioluminescence, fluorescence measurements do not need the presence of a substrate (such as luciferin). Our results provided evidence that bioluminescence methodology has certain advantages over fluorescence measurements of cell lysates in vitro; these advantages include fewer minimum cell numbers (10 vs. 500) and a wider linear signal to cell number (10–106 vs. 103–106 cells) (data not shown). Comparison between luminometer and fluorescence measurements was also performed by linear regression and Bland–Altman analysis, and demonstrated that the two techniques tested are also comparable (Fig. 2C, D). Attenuation of fluorescence was observed for lysates from cells adhered to scaffolds compared to cells alone (control) (Fig. 1C, D). Because the scaffolds tested were not crushed, it is possible that all eGFP from lysed cells adhering to the scaffolds probably was not completely recovered; instead, a portion of protein was trapped and remained inside the material scaffolds. A major drawback of both the fluorescent and DNA quantifications is that they may not be indicative of cell viability but, rather, of total (both live and dead) cell number, because GFP and DNA are relatively stable molecules (half-life >24 h) present in both dead and live cells. In contrast, bioluminescence is indicative of cell viability at the time of assay because of the short-lived (half-life: approximately 2–4 h) activity of the luciferin substrate.25,26 This aspect is a noteworthy advantage for monitoring dynamic cell processes, such as cell proliferation, in real-time.
Moreover, our data provide evidence that the bioluminescence approach under either destructive (cell lysate) or nondestructive conditions facilitates the in vitro assessment of cell number and, thus, expands the scope of the use of bioluminescence to monitoring the fate of transplanted cells for tissue engineering applications. Using a luminometer to measure cell lysate bioluminescence would enable monitoring cell survival and proliferation in material scaffolds in bioreactors in vitro. The bioluminescence procedure using a luminometer is straight-forward, sensitive, easy, and inexpensive, and is a fast technique to implement in the presence of intact scaffolds, but requires cell destruction (lysates). In contrast, IVIS BLI of intact cells enables nondestructive in situ quantification of viable cells within constructs, but it requires an investment in imaging system equipment that is much more expensive than a luminometer.
In vivo, the IVIS imaging system enabled the noninvasive monitoring of the fate of cells present in each scaffold implanted subcutaneously in mice over 59 days. Our results demonstrate that luciferase-expressing cells within scaffolds remain detectable over long periods of time (i.e., 59 days). The ability to noninvasively monitor the in vivo viability and proliferation of cells on 3D porous scaffolds is, undoubtedly, the main advantage of the BLI modality. Thus far, most other studies have relied on invasive methods based on the histological analysis of retrieved implants and required multiple cohorts of animals sacrificed at different time points throughout a study. In contrast, IVIS BLI allows the noninvasive, in vivo, and repetitive collection of images of cells adhering to implanted constructs, enabling longitudinal studies that follow the in vivo performance of individual engineered constructs over prolonged periods of time.
Our measurements provide evidence of large implant-to-implant variability in cell proliferation. Large inter-animal variability of several biological parameters in vivo is a critical issue previously observed by other research groups.19,27 For this reason, the ability to monitor cell viability and proliferation in individual cell constructs in real-time may improve the analysis of the in vivo cell/matrix performance of tissue-engineered constructs. In this respect, the dynamic IVIS BLI technique is unique because it provides quantitative results, specifically, the number of viable cells on the day of implantation as well as the extent of transplanted cell death (70–85% in the present study) occurring within the first 3-day postconstruct implantation; such massive posttransplantation cell death has been widely reported in the literature.8–10,28,29 In contrast to results obtained using bioreactors (Fig. 3), the IVIS BLI revealed opposite trends in vivo cell proliferations on the two scaffolds tested: transplanted cells exhibited a significantly larger (12-fold vs. 2.5-fold; p < 0.005) proliferative index in coral than in AN69 hydrogels (Fig. 6). In our opinion, this case is another example of the difference between in vivo and in vitro results. For this reason, the present study underscored the great usefulness of real-time BLI for screening the performance of scaffold materials regarding to in vivo MSCs survival and proliferation over long periods of time and selecting the most suitable cell construct for tissue engineering applications.
Quantification of viable cells in engineered constructs using BLI has some limitations. First, because of differences in signal attenuation from one scaffold material to another, a comparison of light emissions recorded from different cell constructs should be interpreted with caution unless the results are represented on the basis of normalized data. Second, changes in cell distribution throughout the scaffolds tested as well as degradation of the scaffold material, which may occur in vivo over time, may impact the extent of the attenuation, changing the transmission of the bioluminescence signal compared to the initial signal. Finally, in vivo vascular infiltration and/or extracellular matrix deposition that occur in tissue-engineered constructs over time may also hinder transmission of the bioluminescent signal. For instance, in the present study, postmortem analysis of scaffold integration into the surrounding tissues revealed that all scaffolds were infiltrated by capillaries (data not shown). All of the aforementioned aspects should be taken into consideration when analyzing bioluminescence results. Nevertheless, similar limitations are also encountered when using fluorescence-based imaging techniques. In addition, compared to BLI, eGFP fluorescence is less sensitive, particularly because of its relatively high background signal, which is many times higher than that associated with the luminescent background. 30 For this reason, bioluminescent monitoring of the kinetics of luciferase expression is an attractive method for real-time in situ assessment of cell functions, such as survival and proliferation, in 3D porous scaffolds for tissue engineering and regenerative medicine applications.
Conclusions
The present study established the feasibility of bioluminescence for tracking viable, genetically modified cells distributed within 3D porous scaffolds. Despite an attenuation of the signal when compared to that obtained from cells in suspension, the emitted signal from cell-seeded constructs provided information regarding the cell number present on either soft translucent or hard opaque scaffolds, both in vitro (either from cell lysates or from intact cells) and noninvasively in vivo (subcutaneous implants in mice). Bioluminescence technology is sensitive, easy, and fast-to-implement, and enables noninvasive determination of cell survival and proliferation during prolonged periods of time; for these reasons, bioluminescence technology can be a valuable tool in the tissue engineering and tissue regeneration fields.
Footnotes
Acknowledgments
The authors gratefully acknowledge Dr. Jiri Honiger (Service de chirurgie orthopédique, Saint-Antoine Hospital) for donating the AN69 polymer scaffolds and Inoteb, Inc. for donating the coral scaffolds used in the present study. We would also like to thank the Plate-forme d'Imagerie Dynamique at the Pasteur Institute, Paris, France, for their assistance with bioluminescent imaging and Pr. Rena Bizios for her valuable critical reading of the manuscript. This project was supported by the Centre National de la Recherche Scientifique, the Gueules Cassées Foundation, and the French National Research Agency (ANR) through project MYOCELLOS n° ANR-07-RIB-011.
Disclosure Statement
No competing financial interests exist.