
Abstract
Despite the growing number of clinically practical automated cell culture systems, demand is also increasing for more compact platforms with greater capabilities to prepare primary cells directly from patient tissue. Here we report the development of an automated cell culture system that is also compact. The machinery consisted of a supply unit, an incubation unit, and a collection unit, which fit within a 70 cm × 60 cm × 86 cm space. The compact size was enabled by our concept of using a single culture vessel from the primary culture steps to final cell harvest instead of scaling up with multiple culture vessels. Human fibroblasts and bone marrow stromal cells (BMSCs) were successfully cultured with this system over 19 days without contamination. From three pieces of gingival tissue (2 mm × 2 mm) or from 10 mL of bone marrow aspirate, the system could produce more than 2.0 × 107 cells and up to 3.0 × 107 cells for fibroblasts and BMSCs, respectively. The BMSCs produced by this system were capable of ectopic bone formation after transplantation into the subcutaneous space of nude mice. Our prototype system will provide a foundation for minimizing automatic culture machinery with clinically relevant cell yields while also expanding the automation capabilities to include primary tissue culture.
Introduction
Clinical cell production involves many complicated and experience-based steps, including surgical tissue collection, sample treatments (dissection, dissociation, and dispersion), and cell seeding, which are also known as primary culture. Given the huge individual variation among patients' cells, each of these steps for cell production is made extremely difficult to perform stably using a standardized protocol. In most clinical cases, cell processing for regenerative cell therapy depends largely on the skill and experience of experts. Therefore, significant technological advances are needed for the industrialization of regenerative medicine.
Considering the safety and stability required for cell processing, automation of primary cell culture provides huge advantages for cell therapy. First, in the area of safety, human error, the risk of infectious contamination, or sample cross-contamination could be virtually eliminated. Second, the automation of cell processing promises decreased variability in each operation. Third, as automation hardware becomes more widespread, the operating cost will decrease to less than the cost of hiring skilled technicians and will result in more efficient production of primary cells.
Among reported systems designed for the automation of cell therapy.13–17 Prenosil and Kino-Oka have reported an integrated, computationally operated bioreactor for skin graft production. 16 Their progressive design was based on a solid concept for clinical usage. With that vision in hand, their system was evaluated by testing the ability of their bioreactor to culture keratinocytes that were prepared by a manual primary culture process. Although these results clearly proved that automation of the cell expansion process was effective, the most critical initial steps of establishing the primary culture and the continuous subculture were not automated. Kino-Oka et al. and Terrestege et al. reported incremental advances with their systems supporting continuous subculture.13,14,17 Kino-Oka's work was especially important for demonstrating practical cell expansion with significant reduction of contamination risk by introducing an intelligent subculture system. However, even with these successful improvements in cell culture automation, the primary culture step from tissue was not included.
To design the automatic cell culture system, we believe that a small internal design should be used, which allows the machine to be compact enough to be practical. Laboratory space to place the automatic system is often very limited in clinical facilities, especially in dedicated clean space, such as clean rooms. Also, to accept more patients for treatment, facilities would likely need multiple automatic systems placed in the limited area to perform individualized culture.
In this study, we propose a novel design for subculture automation, which is based on a “single culture flask reuse” concept instead of replacing each individual manual step with a separate mechanized protocol. This idea not only serves to limit the size of equipment but may also have some biological advantages. For example, such a system provides enclosed culture conditions that may reduce the risk of contamination. Further, the reused flask may contain extracellular matrix components produced by the cultured cells, which would enhance cell attachment and proliferation. 18
The performance of this novel design was tested by preparing primary cells directly from human tissues, because this is the most challenging step and not easily substituted with automated culture machines. In this study, fibroblasts and bone marrow stromal cells (BMSCs) were chosen as test models, as they have been already used for clinical purposes by several groups including our own.
Materials and Methods
Cells
This study conformed to the tenets of the Declaration of Helsinki. Primary gingival tissues were obtained from healthy volunteers (26-year-old man and 22-year-old woman) whose informed consent was obtained according to the protocol (detailed in the next section) approved by the ethics committee of Nagoya University Hospital. Primary human BMSCs were obtained from bone marrow aspirated from the iliac crest of three healthy male volunteers (29-year-old, 33-year-old, and 48-year-old men), whose consent was obtained according to the protocol approved by the ethics committee of the Institute of Medical Science, the University of Tokyo (approval no. 16-18-1126).
Tissue preparation and cell seeding
For primary fibroblasts, volunteers' gingival tissue, which is a remnant from a treatment procedure, was used. Mucosal epithelium was removed from the tissue using a scalpel with the aid of a dissecting microscope. The submucosal tissue sample (5 mm diameter) was treated for 1 h at 37°C with 10 mg/mL collagenase (Wako, Osaka, Japan) dissolved in Dulbecco's modified Eagle medium (DMEM) (Invitrogen, Carlsbad, CA) containing 2.5 μg/mL amphotericin B (Sigma-Aldrich, St. Louis, MO) and 50 μg/mL gentamycin sulfate (Sigma-Aldrich). The resulting digested tissues were washed several times in phosphate-buffered saline (PBS; Invitrogen) and resuspended in DMEM containing 10% fetal bovine serum, 2.5 μg/mL amphotericin B, and 50 μg/mL gentamycin sulfate. The suspension of cell aggregates was then injected into the sterile input valve of the apparatus above the cell culture dish using 20 mL syringes each fitted with an 18-G needle. Before injection, the input valve was sterilized with 70% ethanol. With the enzymatic process described above, we found most tissue fragments were digested into cell aggregates. These cell aggregates often retained some of its natural extracellular matrix. Relatively large aggregates usually detached from the dish at an early culture period and were automatically removed at the time of medium change. Smaller aggregates gradually disappeared from subsequent trypsinizations during the subculture process.
For obtaining BMSCs, bone marrow was aspirated from iliac crest under local anesthesia using a syringe containing 3000 U of heparin, diluted with DMEM (Wako) medium up to three fold (30 mL), and injected into the sterile input valve as described above.
Cell culture
Cells were maintained in 150 mL of DMEM with 10% fetal bovine serum, within the culture dish of the incubation unit, and incubated at 37°C, 5% CO2. The incubation unit thermostat module maintained temperature by monitoring temperature changes with a thermosensor. The incubation unit also monitored CO2 using a CO2 sensor, and the incubator atmosphere was maintained at 5% by pumping CO2 from the attached tank. A noncooled black and white CCD camera (MCS40CS; Moswell, Yokohama, Japan) was programmed to continuously record images from multiple fixed positions on the culture dish. Spent medium was discarded by pumping it out to the waste bag connected to the tubing set and fresh medium was then supplied from the supply unit. The media changing functions were part of a job list operated by the control unit personal computer (PC). The medium was kept at 4°C until needed and was warmed up to 37°C during the transfer process before injection to the culture dish.
Cell passage
The timing criteria for cell passage were programmed and executed using the preinstalled software. As there is no historical information for timing criteria with respect to passage of primary cells, the timing was dictated by the total confluence of the culture dish as determined by an operator monitoring via CCD. Individual tasks were programmed into job lists linked to a timetable in the operation. The operation unit automatically controlled execution of the tasks remotely and monitored by an operator. The passage process was carried out using a standard trypsin treatment. This step was done simply using a programmed trypsinization time, but confirmed by an operator in these experiments.
After discarding spent medium and two repeats of washing by PBS, trypsin solution (75 mL) was supplied from the supply unit by rotary pump. The PBS and trypsin solutions kept at 4°C were warmed up to 37°C during the transfer process before injection into the culture dish. After 7 min of incubation, the culture dish of the incubation unit was shaken horizontally several times, until most of the cells in the dish were detached from the dish surface as monitored by the CCD camera. The trypsin reaction was stopped by injecting fresh serum-containing culture medium (150 mL) into the culture vessel.
Passage was performed when the confluence was relatively low. The detached cells were simply dispersed uniformly by shaking the culture dish horizontally, based on the optimized particle dispersion simulation for seeding. This step does not physically extend the culture area, although it helps to disperse the cells evenly inside the culture flask, which is essential to obtain maximum cell number at the time of cell harvest. The detached cells were partially pumped out into the collection tube and the remaining cells were dispersed uniformly in the same culture dish for further processing. The dispersed cells were kept still for about 15 min for reattachment to the dish surface. Shortly after confirming cell reattachment, medium was changed and the cell culture process was repeated. The reattachment step was also automatically performed by the software, but in this experiment it was also verified by an operator.
Cell harvest
When harvesting the cells, the cultured cells were detached from the flask and the trypsinization was stopped, as described in the previous section. The detached cells were then pumped out into the collection tube. When pumping out the cell suspension from the culture dish, the culture dish was inclined by the holder arm to completely collect the solution at the edge of the dish where the aspiration tube was connected.
Cell differentiation
BMSCs were cultured and induced to an osteogenic lineage in the automated culture apparatus. BMSCs were first cultured for 10 days, and then the cells were collected and equally dispersed by trypsinization as described earlier, operated automatically under the job list in the control unit PC. After 4 days of subculture, medium was automatically changed to osteogenic induction medium (DMEM with 10 nM dexamethasone [Sigma-Aldrich] and 100 μM ascorbic acid [Wako]) and induced for 1 week. After the induction period, the cells were harvested into the collecting tube, which was also carried out automatically under the job list in the control unit PC. Osteogenic ability of the cells harvested by the system was estimated by assaying alkaline phosphatase (ALP) activity and ectopic bone forming ability according to a method reported previously. 19 Experiments were done in triplicate for each cell line.
Cell count
Harvested cells were diluted 50-fold with PBS and counted by an automated cell counter CASY Model TT (Schärfe System, Reutlingen, Germany) according to the manufacturer's instructions. Cell viability was also measured by CASY.
Cell transplantation
All experiments involving the use of animals were reviewed and approved by the Institutional Animal Care and Use Committees at the Institute of Medical Science, University of Tokyo. At the day of transplantation, 60 mL of peripheral blood was collected from the donor of primary BMSCs to generate autologous platelet-rich plasma (PRP). PRP was prepared using a kit (Implatex, Tokyo, Japan) according to the manufacturer's protocol. Harvested BMSCs (1 × 106 cells) were mixed with autologous PRP and then mixed with autologous fibrin to form an injectable gel. The gel was mixed with 50 mg of tricalcium phosphate granules (Olympus Terumo Biomaterials, Tokyo, Japan) implanted into the subcutaneous space of 6-week-old female BALB/CAJc1-nu/nu mice (Nihoncrea, Tokyo, Japan). After 4 weeks, the implanted region was surgically collected, stained with hematoxylin and eosin, and observed microscopically (Olympus, Tokyo, Japan).
Results
Development of automated cell culture system
The prototype cell culture platform reported here is comprised of three self-contained components: the supply unit, the incubation unit, and the collection unit (Fig. 1A). This system fits within a footprint of 70 cm × 60 cm × 86 cm and the three main components are linked externally to the operation unit. The supply unit contains a cooler for buffer storage at 4°C and a heater for prewarming reagents to 37°C as they are transferred from the cooled storage area. The collection unit holds the disposable tubes for collecting waste buffers, samples, and cells. The operation unit is a local area network-connected PC with the culture platform operating software running on Windows XP. The operation unit is physically isolated from the cell culture platform so that scientists may run the system remotely.
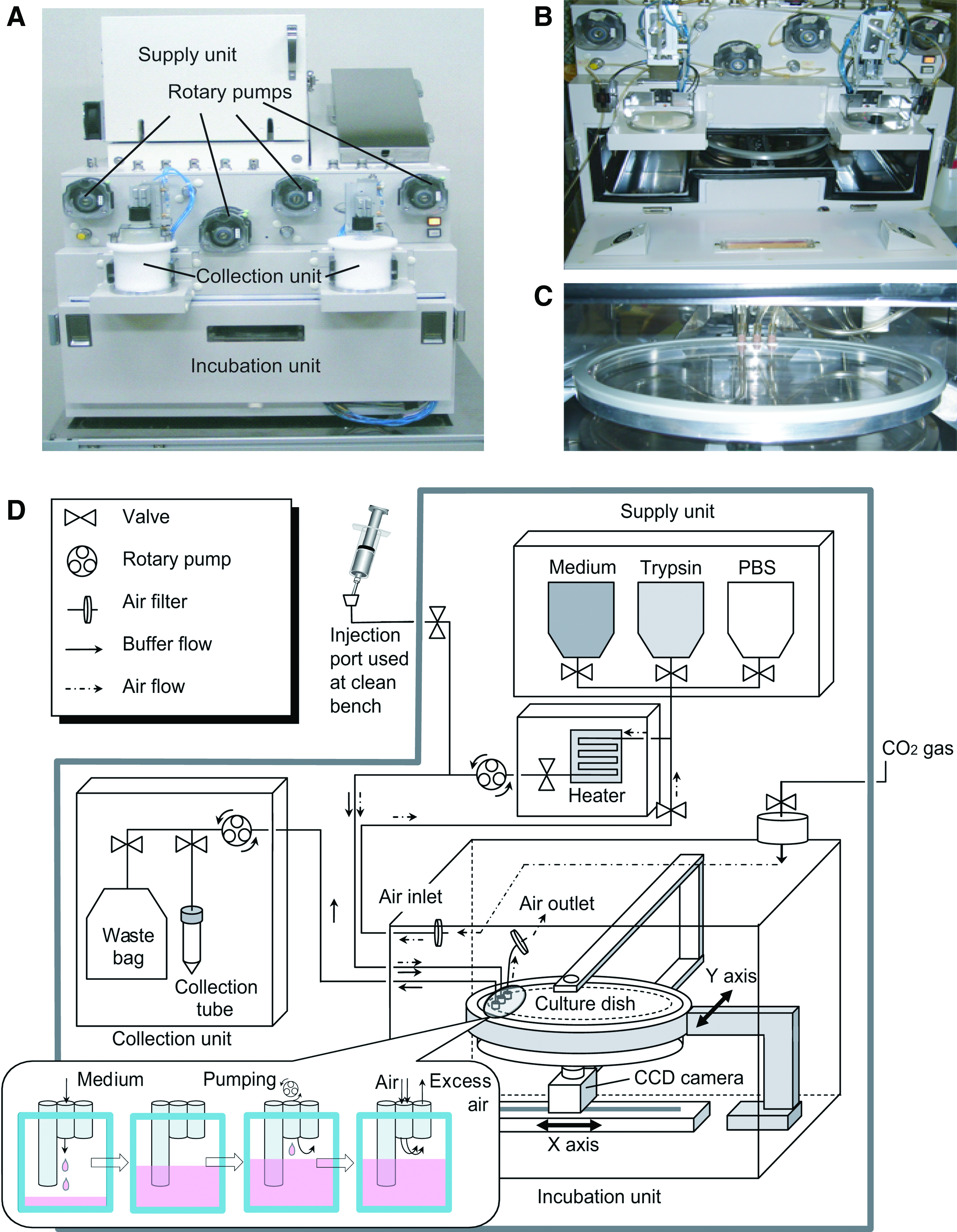
The machinery and the strategy of the constructed automatic cell culture system. (
In the incubation unit (Fig. 1B), the culture dish (Fig. 1C) is placed above the CCD camera onto a movable arm (Fig. 1D) and serves as part of the tubing circuit. The culture dish can be moved and agitated with the actuating arm. The arm can also tilt the culture dish so that the entire volume of buffer or media can be pumped through the connected tubing and into the collection unit. The x/y-axis movement of both the arm and CCD camera allows for real-time imaging of any position on the culture dish. The operation unit's computer controlled all movements of the arm and CCD camera. For the main tubing in the incubation unit, a specialized, sterile, disposable tubing set was used in the culture system. The tubing set includes the culture dish (diameter 25 cm and volume 150 mL), transfer tube, buffer/waste bags (1000 mL each), collection tube, and air filters connected together as an enclosed system. When this disposable circuit is attached to the cell culture platform, the system gains a completely isolated and sterile space for cell culture. Tubing sets were attached to all the pumps and valves in the system by clamping the tubing exterior into the pumps and fittings, which maintained sterility. Using sterile techniques, cells or buffers were injected to prime the tubing on the clean bench before assembly into the automated culture system.
As all of the tubing used in the system has low air permeability and is completely sealed and sterile, the cells in the culture dish require timed aeration. With continuous monitoring of CO2 concentration in the incubation unit, CO2 gas was supplemented to the incubation unit upon demand via a filtered gas tank to maintain a steady state 5% concentration within the system. Both gas and liquid exchange shares parts of a common tubing circuit and flow of gases or liquids is controlled by a series of valves.
CO2 supplementation was accomplished by drawing air (containing 95% CO2) through a 0.22 μm filter (air inlet in Fig. 1D) and into the air tubing circuit, which was connected to the culture medium circuit. To enable air circulation, the valve between the air and liquid tubing circuits is closed and the culture medium rotary pump starts before the valve controlling liquid flow opens. This action creates negative pressure inside the tubing, which prevents liquid from entering the air-only tubing segments together with the valve between the gas and liquid tubing circuit. The fresh air (containing 95% CO2) is pumped through the common tubing circuit and reaches the culture dish via the culture medium inlet. If the CO2 concentration within the incubation unit becomes imbalanced, the sensor and fan will work together to maintain appropriate CO2 levels by adding or exhausting CO2-rich air to the outside of the system.
The inlet connected to the air/medium tubing to the culture dish was designed not to touch the liquid surface of the culture flask. Spent air from the culture flask can passively exit through the 0.22 μm filter to the incubation unit (air outlet in Fig. 1D) without disturbing the medium. A detailed illustration of air/medium injection area attached to the culture vessel in Figure 1D indicates that this design does not aspirate medium during the air exchange process.
A schematic diagram of our system for medium changing and subculture is illustrated in Figure 2. In our system, a single culture dish is placed in the incubation unit. According to the protocol, cells are trypsinized once from the culture dish for harvest and dispersed uniformly in the same culture dish for further processing (see detailed description in Materials and Methods section). We found that clinically useful quantities of cells could be obtained from the automated passage of cells using this system (Tables 1 and 2).

Subculture strategy using a single culture dish. This strategy was introduced to our system to reduce the total platform size. (
FBS, fetal bovine serum.
Control of the entire cell culturing process was performed using the operation unit, which is summarized as a schematic diagram in Figure 3. Tubing set preparation and the analysis of collected cells were carried out manually at a clean bench. With the exception of preparing and attaching the tubing, the user can carry out the entire cell culture procedure automatically, without touching the cell culture dish.
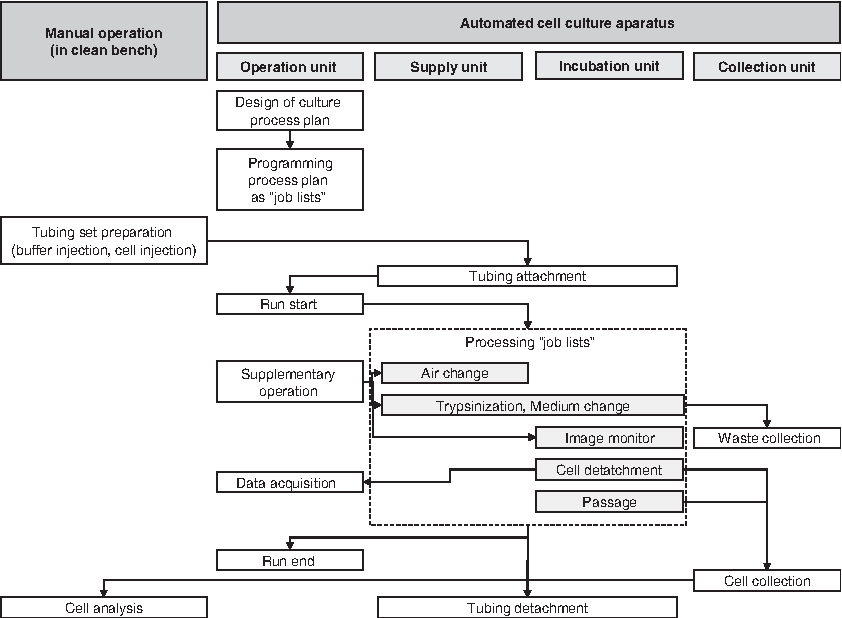
Schematic diagram of automated cell culture procedure. Tubing set preparation and cell analysis are operated manually under sterilized conditions. All the other operations could be controlled from the isolated operation unit using the graphic user interface.
In the operation unit, the graphical user interface provides four windows: (1) main window for all settings, (2) real-time window for obtaining the CCD view, (3) operation window for defining solution conditions (volumes and type), and (4) camera control window for selecting the observation coordinates by clicking the culture dish illustration. The main window can set temperature, CO2 conditions, camera movement, camera focus, and horizontal dish movements. Culture dish images for monitoring the situation could be taken automatically overnight as programmed.
The automated culture system was maintained for a total of four culture cycles (two batches with fibroblasts, and two batches with BMSCs) ranging from 7 to 42 days, with no contamination observed within the system.
Primary culture of fibroblasts using the developed automated cell culture system
Gingival tissues from two different volunteers were partially digested with collagenase and transferred to the machine as a cell and tissue-fragment suspension. The first cell harvest yielded 1.82 × 107 to 2.3 × 107 cells from the three pieces of 2 mm × 2 mm tissue. During the 32 to 35 days of continuous primary culture, all the medium change procedures were executed automatically according to the programmed job list. As shown in Figure 4A, some locally colonized cell growth across several areas of the culture dish was observed after primary tissue seeding. However, after the subculture seeding (Fig. 4B), even cell growth was observed (Fig. 4C). After 7 days of subculture, the cell yield from two volunteers was 2.95 × 107 and 5.0 × 107 cells, respectively (Table 1). This yield was equal to approximately one clinical cell treatment for dermal wrinkle treatment, which requires 1.0 × 107 cells/mL injection.20–22
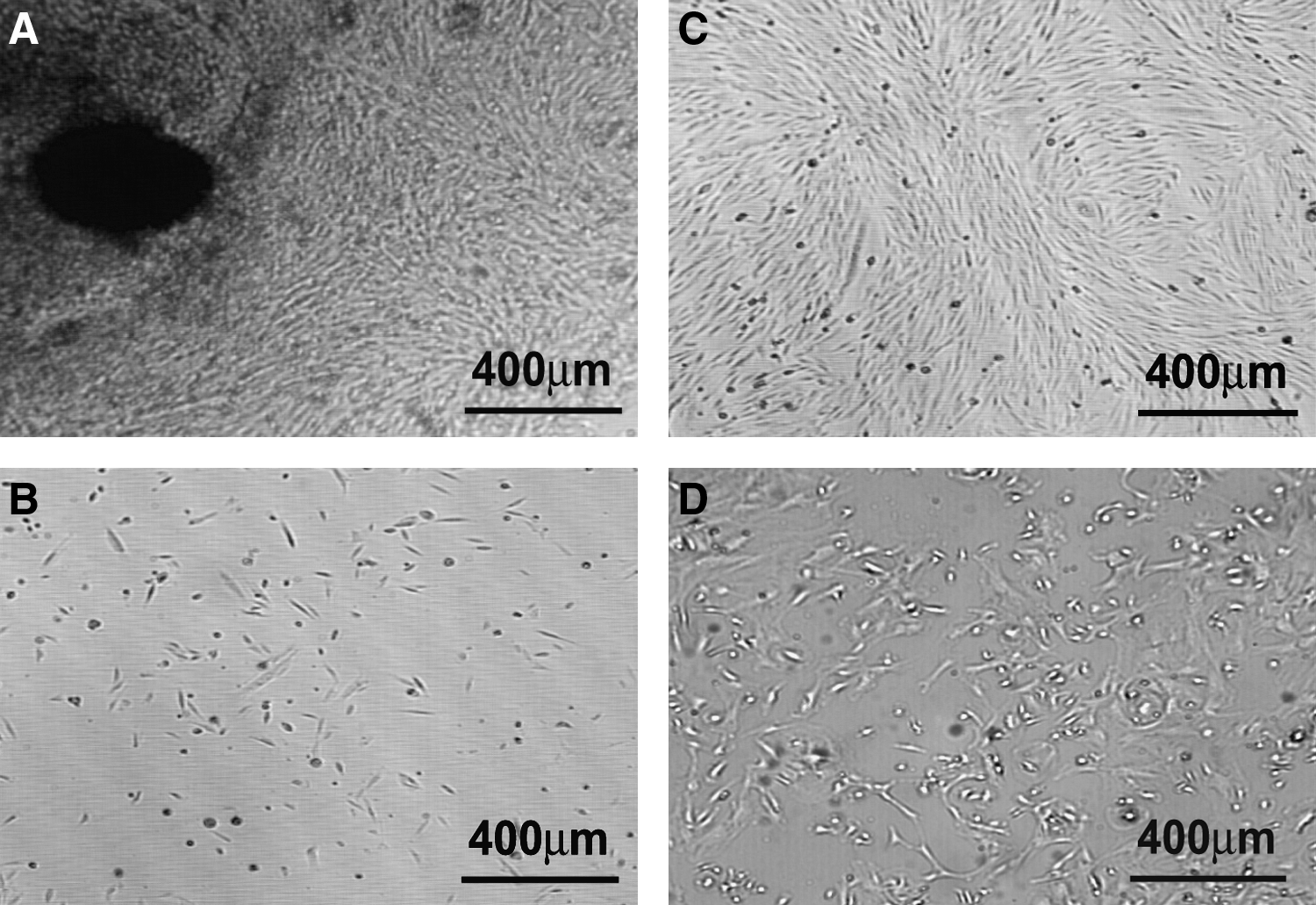
Images obtained from the automatic cell culture system. (
Primary BMSC culture using the developed automated system
To further validate the performance of our system, we cultured primary BMSCs. A bone marrow aspirate from a healthy volunteer was injected into the culture dish after dilution and cultured with preprogrammed culture conditions (Table 2). From the original 10 mL of bone marrow aspirate, 3.0 × 107 cells were obtained after 21 days of culture. Compared with the primary step of fibroblasts, BMSCs were evenly grown (Fig. 4D). For BMSCs cultured in osteogenic induction medium, ALP activity was compared with two other BMSC cultures (from 33-year-old and 48-year-old men) that were manually cultured in T-150 flasks. ALP activity of BMSCs cultured in our system was similar, but lower than that of manually cultured BMSCs (Fig. 5A). Cultured BMSCs from our system were transplanted into the back of nude mice to further test ectopic bone-forming ability. The hematoxylin and eosin-stained sections from the 4-week BMSCs transplants demonstrated clear bone formation, indicating that the cultured BMSCs from our system possessed the differentiation capability necessary for therapy (Fig. 5B).
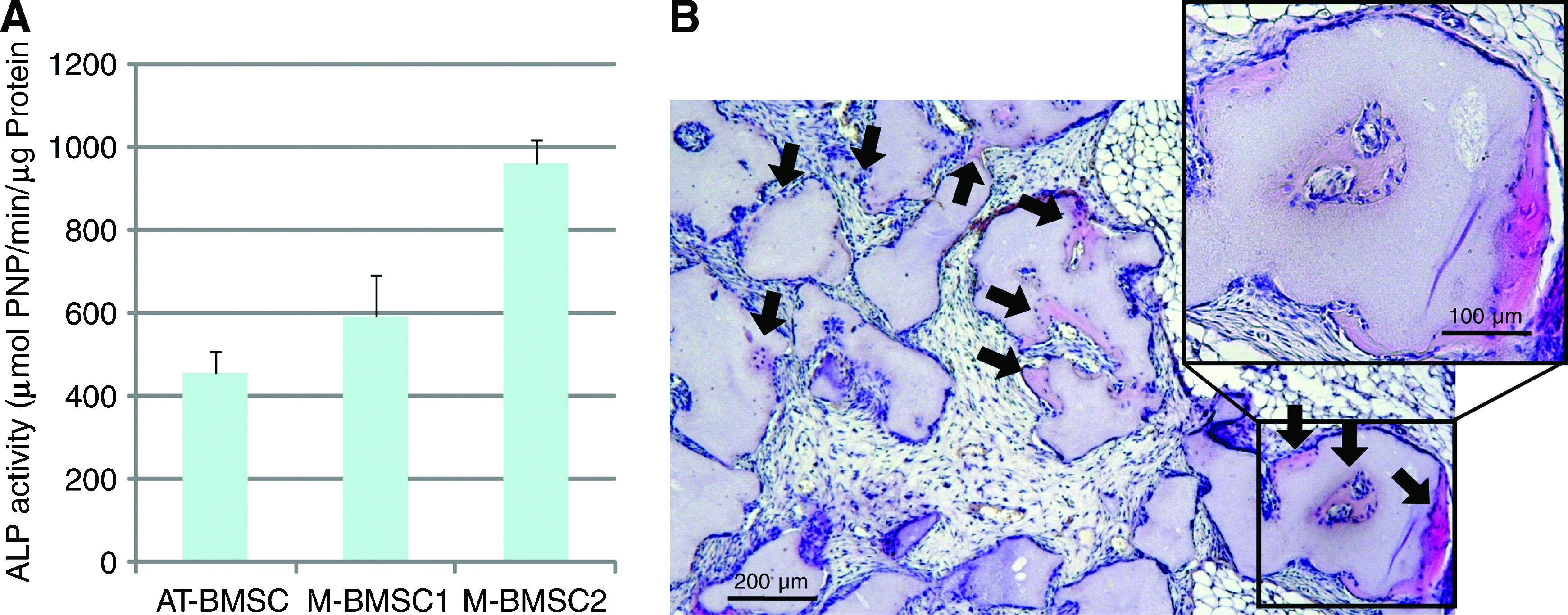
In vitro and in vivo bone-forming ability of BMSCs cultured with the automatic cell culture system. (
Discussion
To satisfy the stringent safety requirements of cell therapy, most of the cell culture processes are operated in a dedicated clean area, such as clean room. Clean rooms are often compactly designed by the limiting expense of construction and maintenance. With clean space at a premium, automation systems must be scaled down to accommodate the increasing demand. Although the size of an automated culture system depends on its function (i.e., single vs. multiple samples) and yield, we were able to scale-down our culture platform by minimizing the size of the incubation unit. Currently, there are reports of existing automated, multifunction culture systems
23
(e.g., larger than 2.0 m × 2.7 m in footprint as indicated at the website;
To develop an automated cell culture system that meets clinical requirements, simultaneous automated primary culture establishment and culture expansion processes are mandatory. The challenge is to perform both of these functions in parallel even though they require separate strategies. Primary culture (i.e., acclimation of target cells) is the step for the cells to transition from their original environment and adjust to the culture environment. With this transition in mind, the culture process should be designed to treat a limited amount of heterogenous cells in a concentrated area. An optimal cell density is usually required for each different cell type to proliferate, and as the number of proliferating cells from primary cultures (including explants) is limited, the early steps should start in a small culture area. 24 In contrast, once the primary cells have been established and the transition from tissue to culture environment has taken place, the objective becomes expansion of those established cells. The expansion process should be designed to promote proliferation of a more homogenous cell population by using an increased culture area, and large vessel size is typically required to obtain clinically useful cell yields. 24
These characteristics of the primary and expansion strategies are conflicting and make automation challenging. If the culture area is too large, the cell density for the primary culture process might be too low to allow the cells to grow, which is especially true under our vessel reuse strategy. In our system, we tried to overcome the incompatible aspects of primary and expansion culture by optimizing cell pretreatment conditions and vessel size.
To test performance optimization, we chose primary fibroblasts and BMSCs as cellular models because they have been used for clinical applications. For BMSC primary culture, bone marrow aspirate, which is already a cell suspension, is simply diluted with culture medium and placed in a culture flask. From an automation perspective, a procedure involving a cell suspension is relatively straightforward, but still requires validation.
In this study, we diluted the bone marrow aspirate in a safety cabinet and injected it into the specially designed vessel, where automated culture would take place. At the primary culture stage, colony formation was observed in limited areas of the culture vessels, which was similar to what has been observed with the manual culture process. 25 With this method, we also obtained sufficient cell yield for clinical application after continuous expansion culture was performed.
For manual fibroblast primary culture, the explant culture technique has been traditionally used. Small tissue fragments are carefully attached to the surface of small culture vessels and covered with small volume of culture medium. 24 From an automation perspective, the explant procedure is difficult to automate, because conventional primary culture techniques use tissue fragments, making it difficult to aspirate and dispense media. Further, the fragment adhesion step is not very robust, leading to loss of primary tissue sample. In this study, we describe optimizing enzymatic disaggregation conditions, which allows for direct injection of digested tissue fragments into culture vessels in the same manner as cell suspensions, facilitating culture automation. From the disaggregated tissues, colony formation was observed in several limited areas of the optimized culture vessel. Despite the large size of the optimized vessel, the observed colonies were similar to those described in other reports of primary culture. 24 Outgrowth from the colonies was observed and yield was found to be sufficient (>1.8 × 107) to start the expansion step.
With both fibroblast and BMSC models, our system was able to evenly distribute the primary cells using the optimized shaking mechanism and replating for the expansion process. The number of recovered cells with our system approximates the number of cells required for a typical clinical cell treatment using fibroblasts.20–22 To obtain a comparable amount of cells using a conventional manual cell culture protocol, 8–10 flasks (225 cm2) would be necessary depending on the condition of the patients' cells.
This culture platform also introduces a monitoring system using a CCD camera to remotely observe the morphology and distribution of the cultured cells from outside the clean cell culture facility. As clean disposable culture tubing and vessels were used and did not require direct contact after the initial setup, remote monitoring avoided the need for additional clean space. This feature contributes to reduce the cost of the cell culture process. In clinical cell expansion, most routine checks are performed based on cell morphology. The real-time/time-lapse function of this monitoring system would be useful not only for the procedural decision-making, but also for quality assurance of the cells. For human cells, large variations have been reported among patients. In the future, real-time monitoring information could be used to individualize culture conditions from patient-to-patient.13,14
The results from this study have clearly demonstrated that our automated culture system, with a single culture vessel, can establish clinical cell cultures and also expand the established primary cells. Successful establishment and expansion of primary cells were accomplished by optimizing pretreatment conditions for primary culture process, vessel size, mechanical optimization, and monitoring of the expansion process.
Conclusions
By culturing different primary cell types with our original system design, we demonstrated the efficacy of a programmed automated cell culture system and the feasibility of introducing such a system into clinical facilities. We are confident that our system concept will contribute to advances in future investigations of automation technologies in regenerative medicine.
Footnotes
Acknowledgments
This work forms part of the Basal Technology Development for the Safety and Efficacy for the Industrialization of Regenerative Medicine (no. 17060601) funded by the Ministry of Health, Labour, and Welfare of Japan. The authors are grateful for the support of Tsutomu Suzuki, Yoshihiro Komori, Yusuke Ando, Hiroshi Tachikui, Hiromichi Shimizu, and Yutaka Wada for establishing the automated system.
Disclosure Statement
The authors declare that no competing financial interests exist in connection with this manuscript.