
Abstract
Laser printing based on laser-induced forward transfer (LIFT) is a new biofabrication technique for the arrangement of biological materials or living cells in well-defined patterns. In the current study, skin cell lines (fibroblasts/keratinocytes) and human mesenchymal stem cells (hMSC) were chosen for laser printing experiments due to their high potential in regeneration of human skin and new application possibilities of stem cell therapy. To evaluate the influence of LIFT on the cells, their survival rate, their proliferation and apoptotic activity, and the DNA damages and modifications of their cell surface markers were assessed and statistically evaluated over several days. The cells survived the transfer procedure with a rate of 98% ± 1% standard error of the mean (skin cells) and 90% ± 10% (hMSC), respectively. All used cell types maintain their ability to proliferate after LIFT. Further, skin cells and hMSC did not show an increase of apoptosis or DNA fragmentation. In addition, the hMSC keep their phenotype as proven by fluorescence activated cell sorting (FACS) analysis. This study demonstrates LIFT as a suitable technique for unharmed computer-controlled positioning of different cell types and a promising tool for future applications in the ex vivo generation of tissue replacements.
Introduction
At present, different techniques, based on ink-jet printing technology4–6 or on laser-induced forward transfer (LIFT), 7 also called laser printing, are under investigation to transfer small amounts of various materials—including living cells—in preset two-dimensional (2D) or 3D patterns. These techniques offer possibilities to control the process with existing computer-aided design and computer-aided manufacturing systems. 8 The ink-jet printing approach is the most commonly used for printing of biological materials.9–11 The important benefit of this technique is the low investment cost, since nearly every ink-jet desktop-printer 9 can be reassembled for the print of biological materials. 12 The main drawback is the very high shear force at the ink-jet nozzle, which leads to significant cell impairment. 13 Therefore, only fluids with low viscosity and low cell density can be printed. Since the laser printing is nozzle free, high viscosity and high cell density solutions can be printed as well. By combining cell solutions with materials that can form stable gels 14 it would further be possible to print 3D tissues layer-by-layer. By using the LIFT approach, living cells have been transferred in a 2D structure.15–19 Further, Barron et al. 20 and Othon et al. 21 transferred single cells also in a 3D structure using layer-by-layer technique.
In the present study, LIFT with gold as a light absorbing layer was investigated to transfer skin cells (fibroblasts/keratinocytes) and human MSCs (hMSC) for the creation of well-defined patterns. Printing of more than one cell type and the generation of multicell patterns were demonstrated. The study was focused on the qualitative as well as quantitative investigation of cell survival, proliferation, apoptosis, and cell DNA damage of the used cell types as well as on phenotype changes of the stem cells after LIFT. The demonstrated laser printing of these cells is a promising step toward the fabrication of living tissue substitutes.
Materials and Methods
Laser printing
The laser printing technique, which is used for defined deposition of cells, is based on the principle of LIFT, as illustrated in Figure 1. The experimental setup consists of two coplanar glass slides. The upper, referred to as “donor-slide,” is covered underneath with a light absorbing gold layer and a layer of the cell containing material to be transferred (Fig. 1). Laser pulses are focused through the glass slide into the gold layer, which is evaporated locally. Laser light absorption in the gold layer generates a high gas pressure that propels the subjacent cell compound toward the lower glass slide, referred to as “collector-slide.” A thin layer of hydrogel or cell medium on the collector-slide provides a humid environment to prevent the cells from dehydration at longer processing times and cushions the impact. Note that the cells survive the impact even without this cushion layer. A hydrogel layer additionally keeps the cells positioned. All transfer experiments in this article are carried out under normal air conditions.
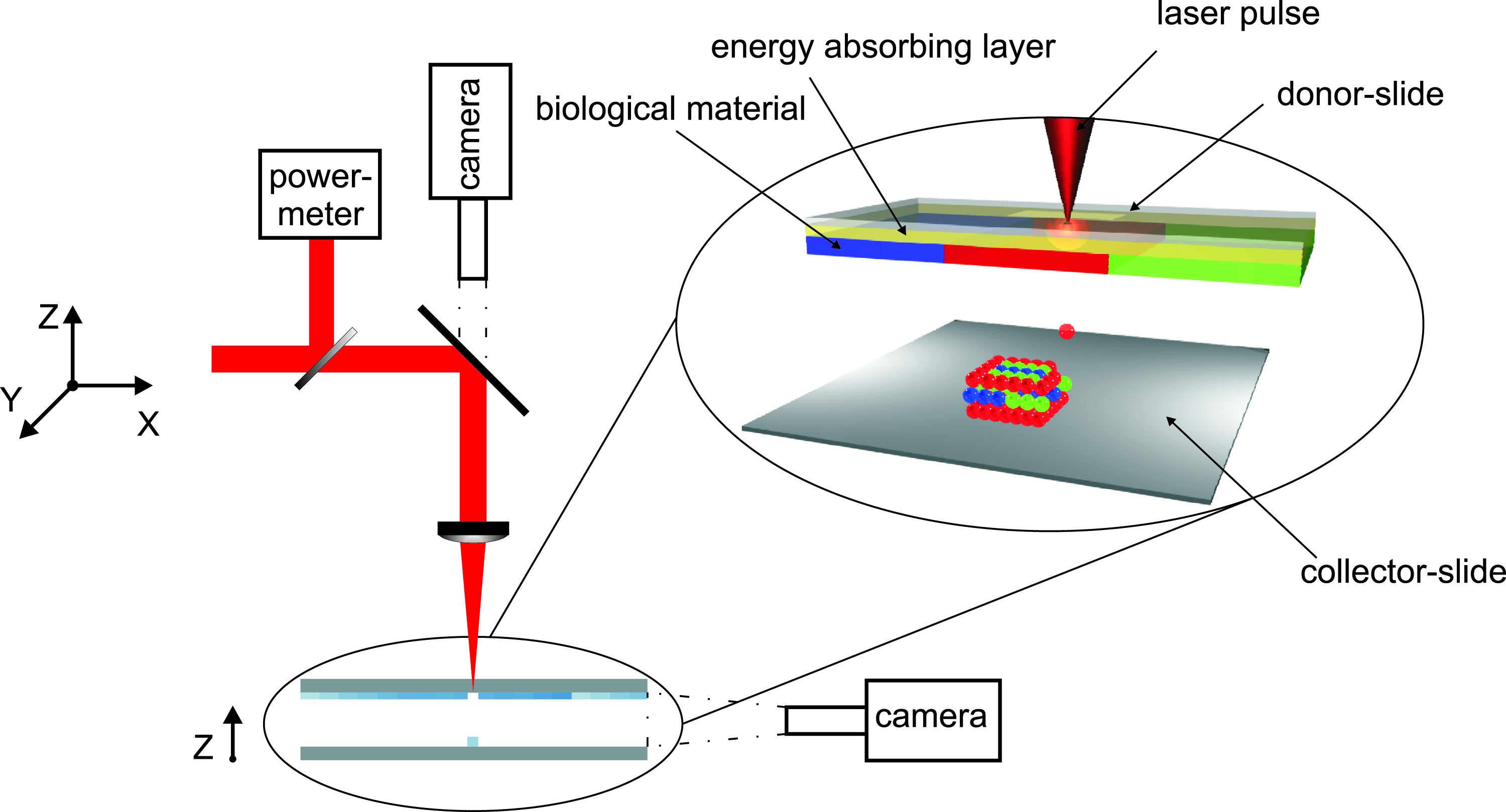
Schematic sketch of the biological laser printing setup and a perspective figure of the principle of laser-induced cell transfer between two coplanar slides: The upper slide is coated underneath with an energy absorbing gold layer and materials to be transferred. Laser pulses are focused through the upper glass slide into the absorbing layer. By evaporating this layer, the material to be transferred is propelled toward the lower glass slide.
The achieved droplet size of the transferred cell compound depends on the laser pulse energy, the gap distance, the focused laser spot size, as well as the thickness of the energy absorbing layer and the cell compound layer. In our experiments, printed droplet sizes with a diameter in the range of 80 to 140 μm are used.
The applied laser is a Nd:YAG-laser (DIVA II; Thales Laser, Orsay, France) with 1064 nm wavelength, 8–9 ns pulse duration (FWHM), and 20 Hz repetition rate. The laser pulses are focused with a 60 mm achromatic lens, producing an ablation spot size of 45 μm in diameter. Depending on the cell/hydrogel layer thickness, the laser pulse energy, measured continuously during the ongoing process, is set between 100 and 190 μJ, corresponding to a laser fluence of 3 to 6 J/cm2.
The cell deposition is controlled via a computerized scanning setup consisting of three high speed translation stages (M-414.1PD and M413.3PD; PI GmbH, Karlsruhe, Germany). On the XY-translation stages, two mirrors are mounted; on the Z-stage holding, the focusing optics as well as the camera for process visualization are mounted. The stages are synchronized with laser pulses over a computer-based real-time system (Adwin-4L-T400; Jaeger Messtechnik, Lorsch, Germany) to ensure equidistant positioning of the laser spots. This automated computer-aided manufacturing controlled stage setup allows the accurate positioning of a wide variety of patterns with a speed of 1200 printed cell droplets per minute, which is only limited by the laser repetition rate applied in this study.
The carrier for the donor-slide is structured modularly to allow quick replacement of the donor-slides coated with different cell compounds. The collector-slide is assembled on a Z-stage to provide controlled positioning with respect to the donor-slide. The gap between the donor- and collector-slide is set to a value in the range of 350 to 500 μm with an accuracy of 5 μm and is monitored through a camera looking parallel to the slides (see Fig. 1).
Preparation of donor- and collector-slides for laser printing experiments
The cultivated cells were trypsinized and centrifuged at 300 × g. Then the supernatant was removed.
For all conducted cell assays, the cell pellet was resuspended in the respective cell medium. This cell suspension was pipetted on the gold-coated donor-slide. The square glass plates with a size of 26 mm and a thickness of 1 mm were coated with a 55 to 60 nm thick gold layer using plasma-enhanced sputter deposition. The cell suspension was dispersed on the gold surface with a blade coater to form a homogenous layer of approximately 50 μm thickness. Then the cell coated donor-slide was fixed in a metal frame and mounted upside-down in the setup. To collect the transferred cells, a sterilized square glass plate (the collector-slide) was mounted parallel to and under the donor-slide. The collector-slide was coated with a layer of cell medium for all the cell assays described here (except for comet assay). This coating was performed with a t-shape spreader. Directly after the LIFT experiments, the laser transferred cells were rinsed from the collector-slide with additional medium into a tube, counted, and distributed to the corresponding well plates for further analysis. The cells that remained on the donor-slide were also rinsed off and collected as control cells. For the analysis of DNA damage, the collector-slide was coated with an agarose layer. The cells printed on the agarose layer were coated with a second layer of agarose and used for the comet assay. For the printing of well-defined cell structures, the cell pellet was resuspended in a mixture of blood plasma and alginate hydrogel at a density of 1–2 × 106 cells in 30 μL. The obtained cell suspension was pipetted on the gold-coated donor-slide and dispersed on the gold surface with a blade coater to form a homogenous layer of approximately 50 μm thickness. To keep the printed cells in their position, the collector-slide was coated with Matrigel™ (BD Biosciences, Heidelberg, Germany). Note that the conducted Live/Dead-assays showed the same results in all studied cases, independent on the materials (cell medium, alginate, blood plasma, Matrigel) used for the preparation of donor- and collector-slides.
Cell culture of skin cell lines
NIH3T3 fibroblasts were cultured in Dulbecco's modified Eagle's medium (DMEM) with high glucose (4.5 g/L) (PAA, Pasching, Austria) supplemented with 10% fetal bovine serum (FBS) (Biochrom, Berlin, Germany), 1% of 100 mM sodium pyruvate (Biochrom), and 1% of 10 mg/mL genta-mycin (Biochrom). HaCaT keratinocytes were grown with DMEM/Ham's F12 medium (PAA) supplemented with 10% FBS and 1% of 10 mg/mL gentamycin.
Isolation and culture of hMSC
Bone marrow–derived hMSC
Collections of bone marrow (BM) aspirates of 12 patients ranging in the age from 68 to 84 years were obtained from the sternum of patients undergoing cardiac surgery at the Cardiac Surgery Department of the University of Rostock. The BM aspirates were received in accordance with the ethical standards of the local ethics committee. The aspirates were diluted 1:5 with 2 mM ethylenediaminetetraacetic acid-phosphate-buffered saline (PBS) (Merck, Whitehouse Station, NY; Nexell, Baxter, Unterschleissheim, Germany). The mononuclear cell fraction was isolated by density gradient centrifugation using Ficoll-Hypaque-Plus solution (GE Healthcare BioSciences, Piscataway, NJ) and seeded at a density of 1 × 106 cells/cm2 into cell culture flasks (Greiner Bio-One, Frickenhausen, Germany). The expansion medium consisted of MSC basal medium (MSCBM; Lonza, Walkersville, MD). Cells were maintained in MSCBM until they reached 70% to 90% confluency. Cells were harvested at subconfluence using Trypsin (PromoCell, Heidelberg, Germany).
Adipose-derived hMSC
Adipose-derived hMSC (Ad-hMSC) were yielded from abdominoplastic surgery performed at the Department of Plastic Surgery at Hannover Medical School with written consent and approval of the ethics committee. Fat lobules were dissected with sterile scissors to lobules of approximately 1 cm3 size. They were washed in equal volumes of sterile Hank's Balanced Salt Solution (HBSS) (PAA), and then their extracellular matrix was digested at 37°C for 1 h with 0.075% collagenase (Biochrom) in PBS under permanent shaking. Then, enzyme activity was neutralized with Hank's BSS containing 10% FBS, centrifuged at 1200 × g for 10 min, and incubated overnight at 37°C/5% CO2 at a density of approximately 150,000/cm2 in a medium (DMEM/Ham's F12) containing 10% FBS, 1% penicillin/streptomycin (Biochrom), and 0.1% ascorbat-2-phosphate. After incubation, the plates were washed with PBS to remove residual nonadherent blood cells. Finally, Ad-hMSC were plated in 150 cm2 cell culture flasks and incubated at 37°C/5% CO2 until they reached 60–70% confluence.
Cell staining
For cell staining, carboxy-fluorescein-diacetate (CFDA) (Invitrogen, Karlsruhe, Germany) and Hoechst 33342 (Invitrogen) were used. Cells were incubated for 15 min at 37°C and subsequently washed thrice in PBS (PAA).
Analysis of cell survival after LIFT
Survival of fibroblasts (NIH3T3), keratinocytes (HaCaT), and hMSC was studied directly after LIFT. Fibroblasts and keratinocytes were shown to be alive or dead after LIFT and this was attained while using the Trypan Exclusion Assay with 0.4% Trypan Blue (Invitrogen). Directly after, the LIFT process cells were rinsed off the collector-slide, counted with a haemocytometer (Fuchs-Rosenthal; Assistent, Sondheim, Germany), and compared to nontransferred cells from the gold layer (control). To evaluate cell membrane integrity of hMSC, fluorescein diacetate (FDA; Invitrogen) and propidium iodide (PI; Carl Roth, Karlsruhe, Germany) were used to differentiate between cells with intact membranes (live; FDA 5.5 μg/mL for 15 min) and membrane damaged cells (dead; PI 50 μg/mL for 15 min). After each stain, cells were washed with PBS, centrifuged, and finally fixed for 10 min with 4% paraformaldehyde for analysis. Live cells were identified by the presence of intracellular, green FDA staining. Red cells, stained by PI, were counted as dead cells.
Analysis of cell proliferation after LIFT
Proliferation of NIH3T3, HaCaT, and hMSC was analyzed using different methods. Proliferation of fibroblasts and keratinocytes was studied using the CellTiter-Blue Kit (Promega, Madison, WI). The test was carried out according to the description of the manufacturer in five replicates per LIFT process. Cells were distributed in a 96-well plate, starting with 500 viable cells in each well and measured with a Genios2 Tecan Reader (Tecan, Grödig, Austria). Proliferation was additionally measured by distributing 1000 transferred vital NIH3T3 cells as well as 860 HaCaT, respectively, in each well of 24-well plates. Over 6 days, every 24 h, 24 chosen samples were trypsinized and stained with Trypan Blue (0.4%) and vital cells were counted separately for each well. Nontransferred cells were used as controls.
To evaluate the influence of LIFT on hMSC proliferation, we assessed the proliferative activity using water-soluble tetrazolium salt (WST-1) assay (Roche Diagnostics, Mannheim, Germany). The WST-1 assay is a colorimetric method in which the dye intensity is proportional to the number of viable cells. HMSC were seeded into 96-well microtiter plates directly after LIFT at a concentration of 2 × 103 cells/well. Cells were then cultured in medium (MSCBM). After incubation of 24 h to 96 h, the cell proliferation reagent WST-1 was added with cell culture medium followed by incubation for 4 h. Sample absorbance was analyzed using a bichromatic enzyme-linked immunosorbent assay reader (Microplate Reader, Model 680; BioRad, Munich, Germany) at 450 nm.
Analysis of apoptosis after LIFT
For determination of apoptosis, fibroblasts, keratinocytes, and hMSC were lysed in ApoOne Buffer (Promega), and N-Acetyl-Asp-Glu-Val-Asp-7-amido-4-methylcoumarin (Sigma-Aldrich, Munich, Germany) was added as a substrate for caspases 3/7. Fluorescence was detected with a Genios2 Tecan Reader. As a positive control, HaCaTs treated for 7 h with 0,525 μM staurosporine (Merck, Darmstadt, Germany) were used.
Analysis of DNA damage through LIFT
To determine whether the LIFT procedure induces genotoxicity effects, a comet assay experiment was performed with fibroblasts (NIH3T3), keratinocytes (HaCaT), and hMSC right after the cell transfer and under control conditions, as previously described. 22 Genotoxicity can be studied by analyzing DNA damages, which are detected with the comet assay method according to single and double DNA strand breaks via a single-cell-gel electrophoresis. These DNA damage effects can be quantitatively described by the parameter tailmoment that characterizes the amount of damaged DNA.
Analysis of hMSC phenotype after LIFT
For characterization of changes in hMSC phenotype, which might be induced by LIFT, cell surface antigen phenotyping of Ad-hMSC was performed 4 days after the laser transfer using flow cytometry. The following cell-surface epitopes were marked with anti-human antibodies: CD90-PE-Cy5 (Becton Dickinson, Heidelberg, Germany), CD29-APC, CD44-FITC (Beckman Coulter, Krefeld, Germany), and CD105-PE (Immunokontakt; AMS Biotechnology, Wiesbaden, Germany). Mouse isotype antibodies served as controls (Becton Dickinson, Beckman Coulter). About 10,000 labeled cells were acquired and analyzed using an FACScan flow cytometer running with CellQuest-Software (Becton Dickinson).
Statistical analysis
To analyze, if a difference between the mean values of corresponding sets of measured values of laser printed cells and controls were real and not only due to statistical variations, Student's t-test (α = 0.05) was performed with the program Origin 8 (Origin Lab, Northampton, MA).
In the case of the used skin cell lines, the “n” refers to the amount of samples in each experiment. The used skin cell lines have always been one strain (e.g., NIH3T3 and HaCaT), as these cells are commercially available cell lines. For the experiments using MSC, the “n” refers to the number of samples used out of 12 patients (1 sample = 1 patient).
Results
Printing of cells
All cells (fibroblasts, keratinocytes, and hMSC) used in this study were transferred successfully by LIFT. A local transfer efficiency of over 90% could be reached, considering the cells in the area wherein laser transfer was performed. In contrast, the overall efficiency also includes cell losses at the blade coater and considers coated areas at the edges of the donor-slide, which are not used for transfer on purpose. Therefore, the overall efficiency is lower than the local transfer efficiency.
Different 2D patterns consisting of one or more cell types were generated. Figure 2 shows a representative immunofluorescence microscopic image of fibroblasts (NIH3T3; green, stained with CFDA) and keratinocytes (HaCaT; blue, Hoechst33342) arranged in a chess board pattern by LIFT. The chess board pattern with a total size of 9.6 × 9.6 mm consists of four lines per square with a line width of 70 μm and a line spacing of 200 μm. This image (Fig. 2) shows the possibility of creating specific patterns consisting of two or even more different cell types, which could precisely be arranged by LIFT.
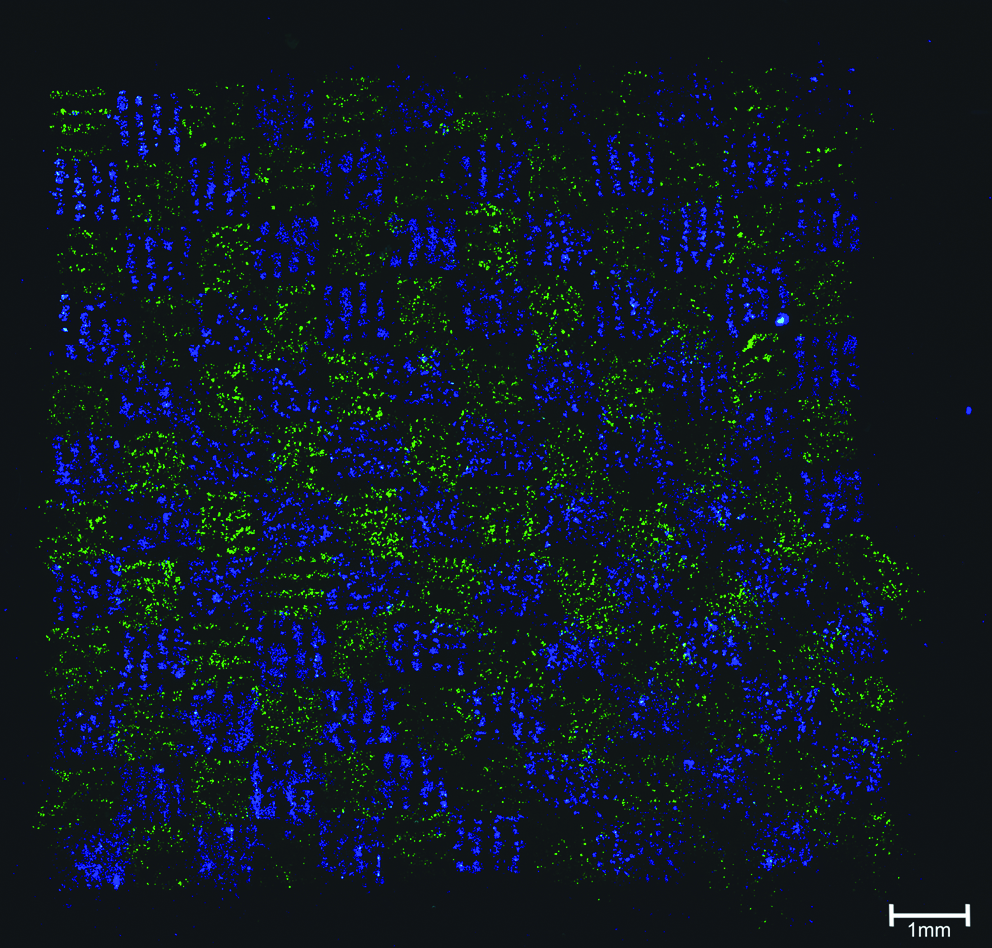
Representative immunofluorescence microscopic image of fibroblasts (NIH3T3; green, stained with carboxy-fluorescein-diacetate (CFDA)) and keratinocytes (HaCaT; blue, Hoechst33342) arranged in a chess board pattern by LIFT. LIFT, laser-induced forward transfer.
Cell survival after LIFT
Survival of cells was determined directly after the transfer. Compared to nontransferred control cells, 98% ± 1% (standard error of the mean [SEM]) of the skin cell lines survived the transfer. The Live/Dead staining of BM–derived hMSC (Bm-hMSC) immediately after transfer showed a survival rate of 90% ± 10% (SEM) versus control.
Cell proliferation after LIFT
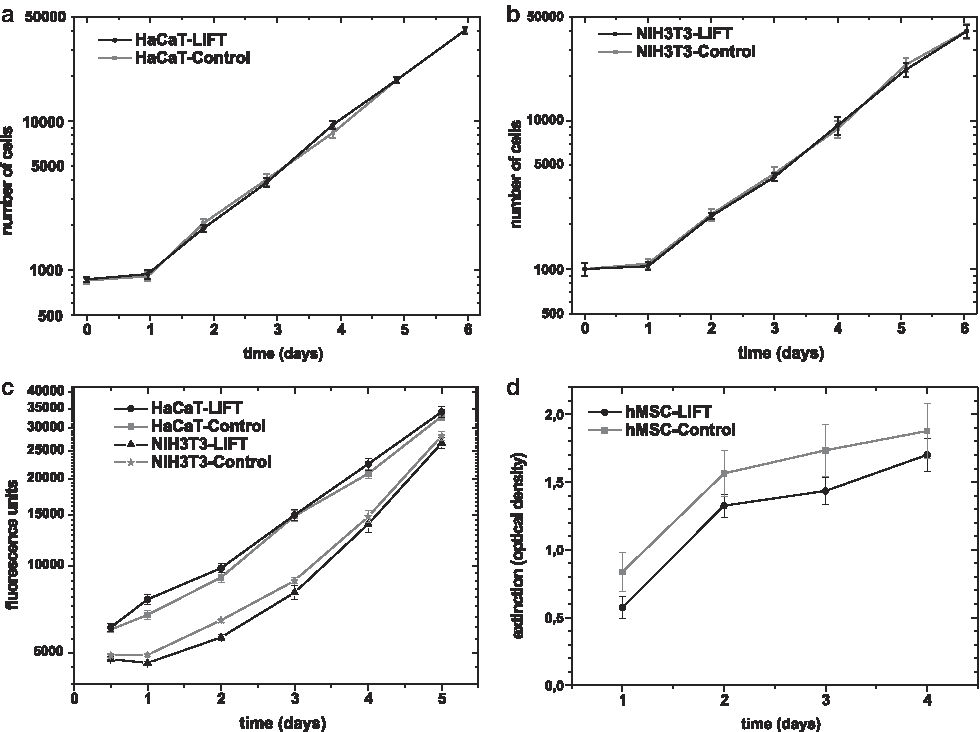
To assess the effect of LIFT on cell growth, we analyzed the proliferation of the transferred cells in comparison to the respective control cells over a time period of several days. Proliferation was measured by two different methods. First, fibroblasts and keratinocytes were directly counted using a haemocytometer. Applying this method (with n = 24 samples each date), we could not detect any major differences in their ability to proliferate after LIFT. Skin cells showed exponential growth starting on day 1 and continuing up to day 6 with no significant differences compared with the control cells (Fig. 3a, b). Second, proliferation was tested by measurement of the metabolic activity, which is proportional to the amount of cells. Using the Celltiter-Blue method for fibroblasts and keratinocytes (each n = 20), transferred skin cells also showed nearly exponential cell proliferation (Fig. 3c), which could already be detected 24 h after LIFT. Albeit significant differences between transferred and control cells were measured at day 1 (fibroblasts, keratinocytes) and two (fibroblasts), the ongoing analysis presented similar curve gradients without statistical significant difference; thus, the difference at the beginning seems to be negligible from the biological point of view.
(
The proliferation of Bm-hMSC, analyzed by WST-1-assay (n = 7), showed a continuously increasing proliferation without significant difference between transferred and nontransferred hMSC (Fig. 3d) until day 4 after LIFT.
Apoptosis of cells after LIFT
Transferred cells were tested for apoptosis as a parameter of possible cell death caused by LIFT. Fibroblasts, keratinocytes, and Bm-hMSC showed no significant differences in the fluorescence signal in the analysis of caspase 3/7 activity between the transferred cells and their respective controls (Fig. 4, each n = 9). This applies for the whole observation period from 12 h up to 48 h after laser transfer.

Assessment of apoptosis of fibroblasts (NIH3T3), keratinocytes (HaCaT), and hMSC, which were printed by LIFT versus nontransferred cells (Control). Apoptosis was assessed by measurement of the activity of caspases 3/7. Values are given as means ± standard error of the mean.
DNA damage through LIFT
Genotoxicity was investigated using the comet assay method. The parameter tailmoment characterizes the amount of damaged DNA. The results are given as mean of tailmoment ± SEM (n = 4). At least 1000 cells per treatment were evaluated. For fibroblasts, keratinocytes, and Bm-hMSC, it could be shown that the LIFT procedure did not significantly increase the incidence of DNA damages. As can be seen in Table 1, the tailmoments of all treatments were comparable with their controls.
At least 1000 cells per treatment were evaluated. The results are given as average of tailmoment as a marker of DNA damages ±standard error of the mean.
LIFT, laser-induced forward transfer; hMSC, human mesenchymal stem cell.
HMSC phenotype
For phenotype characterization of hMSC, surface protein expression of Ad-hMSC from five donors at passage 2 was examined by flow cytometry, 4 days after the laser transfer. Ad-hMSC, similarly to nontransferred control Ad-hMSC, expressed the typical stem cell marker proteins CD44, CD105, CD29, and CD90 without statistical significant difference (Fig. 5). This indicates that LIFT did neither change the immunophenotype of hMSC nor start the initiation of differentiation of stem cells after laser transfer.

Flow cytometric analysis of the phenotype of hMSC. The hMSC expressed the typical MSC-marker proteins CD44, CD105, CD29, and CD90. No significant difference in the ratio of expression between transferred cells and control cells was detected (mean values of %-positive cells ± standard error to the total number of cells analyzed).
Discussion
In the last few years, the ability to print biological material offered a revolutionary new technique for tissue engineering and functional genomics. With these printing techniques, it became possible to create multicellular constructs of arbitrary size with micrometer accuracy. The present study demonstrates LIFT of skin cells as well as hMSC to be a new promising printing method in tissue engineering. The following major findings have been obtained: (i) LIFT is a fast and reliable technique for printing living cells in predefined patterns; (ii) printed cells do not present major differences in survival, proliferation, apoptosis, and DNA-damage compared with respective control cells; and (iii) LIFT does not change the phenotype of the transferred hMSC, thus keeping their high potential for tissue regeneration.
As tissue engineering emerged, it provided many new therapeutic options in regenerative medicine. Besides all the advantages, major problems still exist with the preparation of pure and noncontaminated cells as well as the production of stable 3D-constructs. For this purpose, LIFT might offer the technical precondition for the production of complex 3D tissue-engineered constructs of multiple cell types with high precision, that is, skin or cardiovascular tissue.
In this context, many different skin substitutes have been created, varying in their utilization time (temporary, permanent) and composition (dermal, epidermal, both, with or without cells).23–25 These are already available for clinical use (e.g., Biobrane®, Bertek Pharmaceuticals, Morgantown, WV; Integra®, Integra Life Science, Plainsboro, NJ; Matriderm®, Dr. Suwelack Skin & Health Care, Billerbeck, Germany; Apligraf®, Organogenesis, Canton, MA), offer decreased healing time and less pain, and result in an improved cosmetic outcome.26–28 Regardless, current research still pursues further approaches to improve the quality of skin substitutes. Therefore, LIFT might offer a new procedure for the fast, precise, and reliable production of a ready-to-use skin equivalent.
The laser printing of fibroblasts, keratinocytes, and hMSC demonstrated high survival rates as one of the most important preconditions of this technique. Additionally, the assessment of cellular proliferation by two different methods and the possible induction of apoptosis studied by ApoOne did not present any biological significant changes compared with the respective control cells, which have not been transferred by LIFT.
Similarly to the used skin cells, laser transfer of hMSC also was studied and evaluated for the same parameters. MSC are found in many adult tissues and represent an attractive stem cell pool due to their self renewing ability, high differentiation capacity, and mesodermal differentiation potential. 29 Therefore, MSC are thought to regenerate many human tissues like bone, cartilage, adipose tissue, skin, cardiomyocytes, and even insulin producing tissue. 30 The printing of hMSC with a high survival rate demonstrates LIFT to be a very promising method for stem cell therapy. We could also not detect any differences in proliferation and apoptosis for hMSC. In addition, flow cytometric analysis demonstrated that LIFT does not induce differentiation of hMSC so that stem cells could be transferred safely.
In conclusion, the present study proved LIFT to be a reliable and safe technique for the unharmed computer-controlled positioning of different cell types. The standardized and statistical evaluation, as shown here, is an important precondition for the ongoing research in laser printed tissue-engineered constructs. Therefore, LIFT is a promising tool for future applications in the ex vivo generation of tissue replacements. The 3D printing of skin equivalents for burn patients or the production of cardiac tissue or even cardiac valves might be potential areas of application.
Footnotes
Acknowledgments
The authors kindly thank Mrs. M. Fritsche from the Department of Cardiac Surgery, University of Rostock, and Mrs. U. Fuhrmann and Mr. H. Naghilouy Hidaji from the Department of Plastic, Hand, and Reconstructive Surgery, Hannover Medical School, for their excellent technical assistance and organizational help.
This study has been supported by Deutsche Forschungsgemeinschaft, SFB TransRegio 37 and Rebirth Cluster of Excellence.
Disclosure Statement
No competing financial interests exist.