
Abstract
Mesenchymal stem cells (MSCs) are capable of self-renewal and differentiation along the osteogenic, chondrogenic, and adipogenic lineages and have potential applications in a range of therapies. MSCs can be cultured as monolayers on tissue culture plastic, but there are indications that they lose cell-specific properties with time in vitro and so poorly reflect in vivo MSC behavior. We developed dynamic three-dimensional (3D) techniques for in vitro MSC culture using spinner flasks and a rotating wall vessel bioreactor. We characterized the two methods for dynamic 3D MSC culture and compared the properties of these cultures with monolayer MSCs. Our results showed that under optimal conditions, MSCs form compact cellular spheroids and remain viable in dynamic 3D culture. We demonstrated altered cell size and surface antigen expression together with enhanced osteogenic and adipogenic differentiation potential in MSCs from dynamic 3D conditions. By microarray analysis of monolayer and spinner flask MSCs, we identified many differences in gene expression, including those confirming widespread changes to the cellular architecture and extracellular matrix. The upregulation of interleukin 24 in dynamic 3D cultures was shown to selectively impair the viability of prostate cancer cells cultured in medium conditioned by dynamic 3D MSCs. Overall, this work suggests a novel therapeutic application for dynamic 3D MSCs and demonstrates that these methods are a viable alternative to monolayer techniques and may prove beneficial for retaining MSC properties in vitro.
Introduction
MSCs are commonly cultured as monolayers using conventional tissue culture techniques. These methods have proved adequate but there are reports demonstrating a loss of the replicative ability, colony-forming efficiency, and differentiation capacity of MSCs with time in culture.1–3 Together with the fact that MSCs comprise only a small proportion of the bone marrow and must be extensively expanded in culture to achieve the numbers required for any therapeutic strategy, this highlights the requirement for improved methods for the in vitro culture of MSCs.
The application of three-dimensional (3D) cell culture techniques is receiving increased interest with evidence showing significant differences between the cellular phenotype and biological response of cells cultured in monolayer and 3D cell culture. 4 The 3D methods facilitate greater cell–cell contacts and interactions of cells with the extracellular matrix (ECM), allowing cells to adapt to their native morphology, which may influence signalling activity. As a result, it is becoming increasingly accepted that 3D culture methods provide a cellular environment more consistent with that in vivo. Approaches providing a 3D environment have included the use of porous scaffolds, collagen gels, and multicellular aggregates,5,6 each of which have been successfully employed with a variety of cell types.7–12 Cancer biology, in particular, has been transformed by the use of 3D techniques as these can more accurately reproduce the response of cells in a tumor. 13 Despite these positive results, the use of 3D methods is still in its infancy and it is likely that such techniques will prove beneficial in many more cases, particularly the stem cell field where the cellular microenvironment is known to have a profound influence on the biology of the cells within it. 14 There is also a growing interest in the use of bioreactors to provide a dynamic culture environment that will increase the flow of oxygen and nutrients to cells and the removal of waste products from cells and produce forces such as fluid shear stress, which confer biomechanical cues. Such forces constitute another important aspect of the cellular environment and can influence the properties and behavior of cell cultures including MSCs.15–18
We have used spinner flasks and a rotating wall vessel (RWV) bioreactor to culture MSCs under 3D, dynamic conditions, and both these systems have been successfully used to culture a number of cell types and tissue constructs.19–22 Spinner flasks have an internal magnetic stirring arm and two side-arms that can be used for the addition or removal of agents and to provide aeration to the contents of the flask. The flasks are cultured on a stirring platform which is used to control the speed of the internal arm. The RWV bioreactor consists of a cylindrical culture chamber that contains an internal, membrane-covered cylinder through which oxygen is drawn into the vessel as it rotates. When in use, the bioreactor is completely filled with culture medium and the medium rotates as a solid body. Both systems provide a means of mixing the culture medium and are often used to culture larger tissue constructs in which the mixing of the culture medium helps to provide a flow of oxygen and nutrients to the cells in the construct. However, the absence of air pockets in the RWV and the diffusion of oxygen and carbon dioxide through a membrane ensures that there are no bubbles in the culture medium and provides conditions with reduced turbulence and fluid shear stress when compared with spinner flask cultures.
Here, we characterized MSCs from spinner flask and RWV cultures and demonstrated that these methods are a viable alternative for the in vitro culture of MSCs. In addition we show that there are fundamental differences between the cells cultured in monolayer and dynamic 3D cultures, that is, altered immunophenotype, including loss of human leukocyte antigen (HLA) class I molecules, and enhanced osteogenic and adipogenic differentiation in MSCs cultured using the dynamic 3D techniques when compared with monolayers. Microarray analysis allowed us to investigate these differences further. The data confirmed the basis of many of the morphological changes observed and provided new insights into MSC biology. In addition, the upregulation of interleukin 24 (IL24) by dynamic 3D MSCs was determined to impair the viability of a number of pancreatic cancer cell types while leaving noncancer cells unaffected, thereby providing a novel way by which dynamic 3D MSC cultures could be used for future cancer therapies.
Materials and Methods
All plasticware were obtained from Corning (Artington, UK) and reagents were purchased from Invitrogen (Paisely, UK) unless otherwise stated. Cells were cultured at 37°C in a humidified atmosphere with 5% carbon dioxide.
Cell culture
Primary human MSCs were isolated from femoral heads, after obtaining informed consent from the patients of Harrogate District Hospital during routine hip replacement surgery as previously described.
23
Briefly, trabecular bone was removed from the femoral head and collected in α-minimum essential medium with 100 units/mL penicillin and 100 μg/mL streptomycin (αMEM/ps). The trabecular bone was minced, the supernatant was collected, and cells were pelleted. The resuspended cells were passed through a 70-μm cell strainer (BD Biosciences, Oxford, United Kingdom) before separation on a Ficoll-Paque Plus (Amersham Biosciences, Little Chalfont, United Kingdom) density gradient. The white mononuclear fraction was harvested, and the cells were washed with 5 mM ethylene diamine tetra-acetic acid, 0.2% bovine serum albumin in phosphate-buffered saline before seeding into a tissue culture flask with αMEM + ps supplemented with 15% batch-tested fetal bovine serum (FBS). MSCs were expanded in T175 tissue culture flasks with αMEM/ps + 15% FBS. To promote spheroid formation, MSCs were incubated for 6 h at a density of 1 million cells/mL in a nonadherent plate, dispersed into smaller aggregates by gentle pipetting, and transferred to either spinner flask (Techne, Staffordshire, United Kingdom) or RWV (Synthecon, Bereldange, Luxembourg). Culture medium was added to give an initial cell density of 20,000 cells/mL. The MSC aggregates (spheroids) were then cultured with a stirring speed of 30 rpm for the spinner flask and a rotation speed of 15 rpm for the RWV. To promote differentiation, medium supplemented with 15% heat-inactivated FBS and osteogenic or adipogenic factors was added upon transfer of cells to the dynamic cultures or to parallel monolayer cultures at a density of 10,000 cells/cm2 and fully exchanged at 7-day intervals. Osteogenic differentiation was induced with 50 μg/mL
C3H10T1/2 and SaOS-2 cells were obtained from the (American Type Culture Collection, Manassas, VA), and prostate cell lines were kindly provided by Prof. Normal Maitland and Dr. Anne Collins (University of York). C3H10T1/2, PNT-1a, PNT2-C2, LNCAP, VCAP, PC3, and DU145 cell lines were cultured in Dulbecco's modified Eagle's medium and SaOS-2 cells in αMEM, both supplemented with 1% ps and 10% FBS. Human umbilical vein endothelial cells grown in endothelial cell growth medium/SupplementMix (Promocell, Heidelberg, Germany) supplemented with 10% FBS and 1% ps were purchased.
Histological analysis of spheroids
MSCs were cultured as spheroids in spinner flasks and RWV for 7 days, fixed in 10% formalin, dehydrated through a graded series of alcohols, and incubated in Histoclear II (Fisher Scientific, Loughborough, United Kingdom) before being transferred into molten wax. Five-micrometer sections were cut using a (Leica, Milton Keynes, United Kingdom) RM2165 rotary microtome and stained with hematoxylin and eosin.
Scanning electron microscopy
The samples for scanning electron microscopy (SEM) were fixed in 2.5% gluteraldehyde, dehydrated through a graded series of ethanols, incubated for a further 20 min in hexamethyldisilazane (Sigma), and dried overnight. The samples were mounted onto carbon-coated chucks, sputter-coated with gold in an argon atmosphere for 2 min at 1 kV, and analyzed on a (Jeol, Welwyn Garden City, United Kingdom) 6490LV scanning electron microscope.
Transmission electron microscopy
The samples for transmission electron microscopy (TEM) were fixed in 2.5% gluteraldehyde, then in 1% OsO4, dehydrated through a graded series of ethanols, incubated in epoxy-propane for 25 min, infiltrated with epon araldite, and polymerized for 48 h at 60°C. Sections of 70 nm were cut, stained in saturated uranyl acetate in 50% ethanol and Reynolds lead citrate and analyzed on a (FEI Technai, Eindhoven, The Netherlands) Tecnai G2 transmission electron microscope. Representative images were taken for two independent donors.
Live/dead viability assay
After 7 days of culture, cell viability was determined using the live/dead reduced biohazard viability/cytotoxicity kit (Molecular Probes, Paisley, United Kingdom) according to the manufacturer's instructions. The samples were imaged using a Carl Zeiss Ltd, Welwyn Garden City, United Kingdom, LSM 510 meta on an Axiovert 200 M microscope and images were taken as a z-series through to the center of the spheroid.
Flow cytometry
MSCs were harvested by trypsinization and passed through a graded series of needles to produce a single-cell suspension. Monolayer and spheroid samples were treated in an identical manner. The cells were resuspended in wash buffer (phosphate-buffered saline containing 5 mM ethylene diamine tetra-acetic acid and 0.2% bovine serum albumin). To determine cell cycle stage, the samples were analyzed after the addition of 2 μg/mL 4'6-diamidino-2-phenylindole for 10 min. To determine cell surface antigen expression, the samples were incubated with primary antibody (mouse monoclonal anti-CD29, CD34, CD44, CD45, CD73, CD90,CD105, CD166 from (Beckton Dickinson, Oxford, United Kingdom) and HLA class I from Chemicon International, Chandlers Ford, United Kingdom), washed with wash buffer, and with the exception of the anti-CD166 antibody which was directly conjugated to phycoerythrin (PE), incubated with a fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse antibody (Sigma). Isotype controls were incubated with the same concentration of mouse IgG (Sigma Aldrich) instead of primary antibody. The samples were analyzed using a (Dako Cytomation, Ely, United Kingdom) CyAn flow cytometer which counts 10,000 cells per sample. Positive cells were determined as the proportion of the population with higher fluorescence than 95% of the isotype control. Representative images were taken for two independent donors.
Alkaline phosphatase and von Kossa staining
Alkaline phosphatase activity was detected with 1 mg/mL Fast Red-TR (Sigma) and 0.2 mg/mL Napthol AS-MX Phosphate (Sigma) in 0.1 M Tris-HCl (pH 9.2) for 2 min at room temperature. von Kossa-positive mineralization was detected by a 30-min incubation in 1% silver nitrate, followed by 5 min in 2.5% sodium thiosulphate.
Oil Red O staining
Lipid droplets were detected by fixation for 5 min in 60% isopropanol and staining for 30 min in 60% Oil Red O in water (from a 0.5% Oil Red O in isopropanol stock; Sigma).
Quantitative real-time polymerase chain reaction
Quantitative real-time polymerase chain reaction (qRT-PCR) was carried out using the ABI Prism 7000 (Applied Biosystems, Warrington, United Kingdom) sequence detection system with SYBR green chemistry. The samples were run in triplicate and experiments were repeated using three independent donors. The results from one representative donor are shown.
Total RNA was extracted using TRIzol (Invitrogen, Paisley, United Kingdom) according to the manufacturer's instructions and treated using the DNA-free kit from (Ambion, Warrington, United Kingdom). cDNA was synthesized from 300 ng total RNA with 200 U SuperScript II, or the equivalent volume of DNase and RNase-free water for no-reverse transcriptase (RT) controls, and any residual RNA degraded with RNase H. Twenty-five microliters of reaction mixture contained 1 × SYBR green master mix (Applied Biosystems, Warrington, United Kingdom), 0.8 μM forward and reverse primer, 50 ng cDNA, and DNase and RNase-free water. Thermal cycling conditions were 2 min at 50°C and 10 min at 95°C, followed by 40 cycles of 15 s at 95°C and 1 min at 60°C. Data were analyzed by the 2−ΔΔCt method using RPS27a as a housekeeping gene and were displayed as log2 mean relative expression ± standard error of the mean. Data were analyzed by t-test with unequal variance.
Affymetrix analysis
MSCs from three independent donors were cultured as monolayers or spinner flask spheroids for 7 days in basal MSC medium before isolation of RNA using TRIzol. The samples were treated with DNase I for 1 h at 37°C and the DNase was inhibited by addition of 1 μL ethylene glycol tetra acetic acid (EGTA) for 10 min at 65°C. RNA quality was determined using an (Agilent Technologies, Wokingham, United Kingdom) 2100 bioanalyzer and only samples with a RNA integration number greater than 9.0 were determined to be of sufficiently high quality to proceed. Sample processing and array hybridization were performed in the Genomics Laboratory of the Technology Facility at the University of York, using 1 μg total RNA in a one-cycle target labeling method. The samples were hybridized to Affymetrix Human Genome U133 Plus 2.0 arrays. The raw data were processed by the resident bioinformatician using ArrayAssist (Stratagene, Amsterdam, The Netherlands) with background correction, data normalization, and probe summarization performed using the MAS5 algorithm. Statistically significant changes in gene expression were determined by independent t-test for all genes present in at least one out of the six samples. ArrayAssist was also used to examine the distribution of the data and to assign and probe gene ontology (GO) terms using all GO terms where p ≤ 1.
Conditioned medium and MTT assays
Conditioned medium was obtained from at least three MSC donors by culturing MSCs as monolayers or spheroids at equal cell densities in basal MSC medium. After 7 days the culture medium was collected, centrifuged for 5 min at 450 g, and passed through a 2-μm filter. This conditioned medium was used to treat cell lines plated at a density of 5000 cells/well in a 96-well plate. For blocking experiments, increasing concentrations of an antibody with known IL24-blocking activity (200 ng/mL; R&D Systems, Abingdon, United Kingdom) was added to LNCAP and PNT-1a cells treated with 3D conditioned medium. After 4 days, 30 μL of 5 mg/mL MTT [1-(4,5-dimethylthiazol-2-yl)-3,5-diphenylformazan; Sigma] was added to each well for 3 h at 37°C before the addition of 100 μL lysis buffer (0.1 M HCl in isopropanol) for 1 h at 37°C. Absorbance was measured on a Dynex MRXII microplate reader (Dynatech, Billinghurst, United Kingdom) at 570 nm. The data were normally distributed with equal variance, as determined by Kolmogorov–Smirnov and Levene's tests, and were analyzed by one-way analysis of variance with Tukey's post hoc test.
Results
MSCs form 3D multicellular spheroids when cultured in dynamic conditions
MSCs were cultured under dynamic conditions in spinner flask or RWV culture for 7 days. Under optimized cell number and growth conditions, the cells formed compact multicellular spheroids. Following histological analysis, we identified a densely packed structure with cells present through to the center of the spheroid, with no evidence of a necrotic core (Fig. 1). Measurements of the spheroids determined an average diameter of 98.7 μm for spinner flask and 31.7 μm for RWV spheroids, with the size of spinner spheroids ranging between 56.1 and 135.4 μm and those from RWV culture between 18.1 and 43.5 μm (n = 10).

MSCs aggregate to form compact multicellular spheroids under dynamic culture conditions. Phase contrast micrographs and hematoxylin and eosin (H + E)-stained sections from spinner flask and RWV spheroids after 7 days of dynamic culture. Scale bars = 20 μm. MSCs, mesenchymal stem cells; RWV, rotating wall vessel.
To follow spheroid formation during this 7-day period, SEM and TEM were used to analyze the surface characteristics and internal organization of spinner flask and RWV spheroids after 1, 3, and 7 days of dynamic culture. Using SEM we identified an irregular topology with protruding cell bodies on spheroids on day 1 and to a lesser extent on day 3. By day 7, MSCs in both spinner and RWV cultures compacted more effectively to form regular spheroidal structures with flattened surfaces (Fig. 2A). Higher magnification images revealed that cells on the surface of the spheroids became progressively more spread up to day 7 when they formed a flattened layer on the surface of the spheroid with the boundaries between separate cells becoming less distinct (Fig. 2B). This observation was confirmed by TEM analyses which demonstrated that a contiguous arrangement of elongated MSCs develops on the outer surface of the spheroids by day 7. MSCs from the spheroid interior had a more irregular morphology (Fig. 2C).

Scanning electron microscopy (SEM) and transmission electron microscopy analyses of spheroid formation. (
MSCs remain viable and proliferate in dynamic 3D culture
The viability of the cells within the spheroids was determined by live/dead assay after 7 days of dynamic culture. The majority of the cells were viable in spheroids from both spinner flask and RWV cultures, with no apparent differences between cells on the periphery and those in the center of the spheroid. The overall cell viability remained comparable to MSCs cultured as a monolayer (Fig. 3A). Many methods commonly used to determine cell proliferation rates are incompatible with the spheroid system and so flow cytometric analysis of cellular DNA content was used to provide a snapshot of the number of cycling MSCs after 7 days of culture. The proportion of cycling cells was determined to be 3.9% for monolayer MSCs and 4.3% or 3.5% for spinner and RWV MSCs, respectively, indicating an equivalent rate of turnover (Fig. 3B).
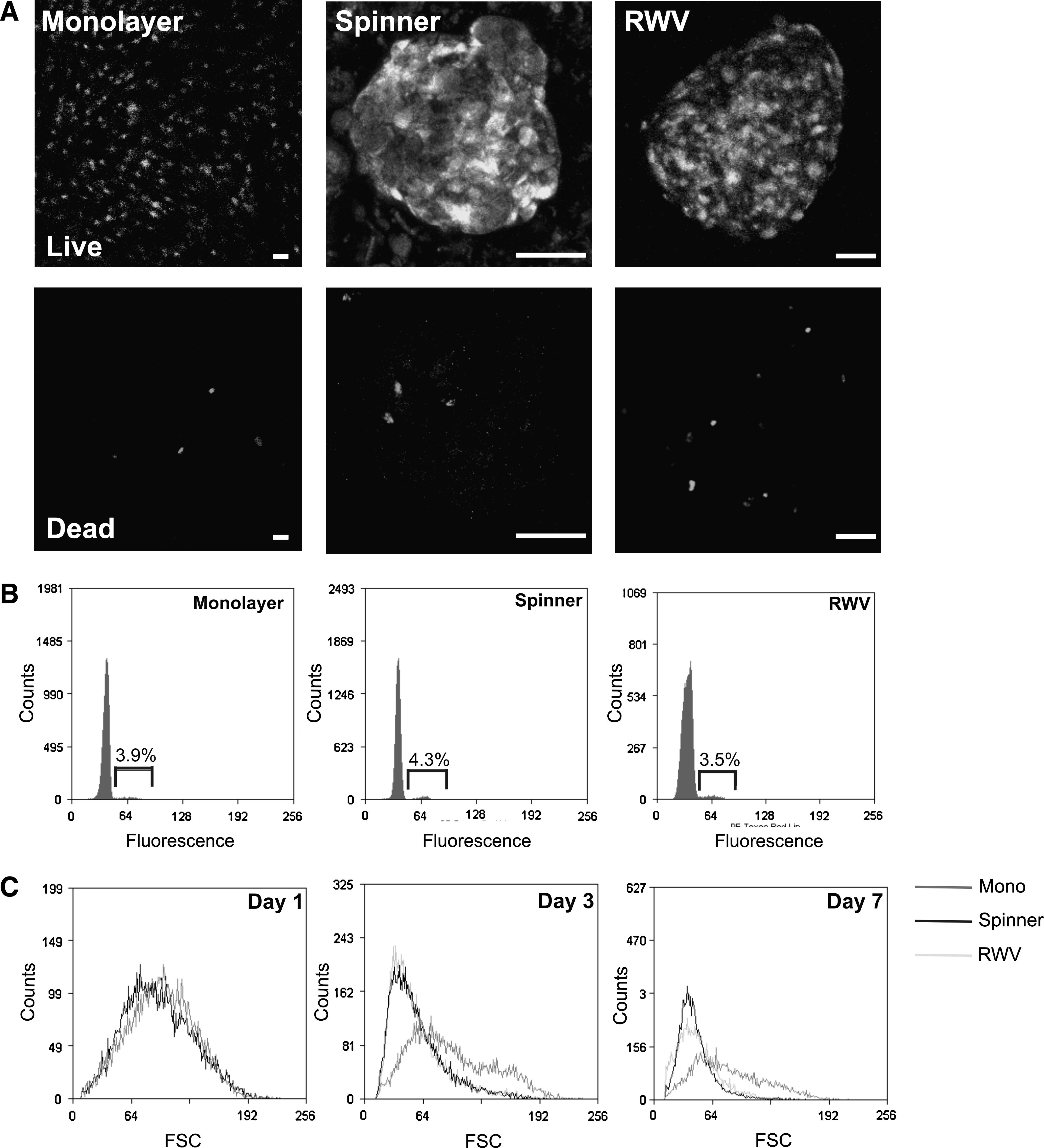
MSCs remain viable and proliferate in dynamic 3D culture conditions. (
Cell surface antigen expression of MSCs changes under dynamic 3D culture conditions
We analyzed the expression of a panel of MSC surface antigens in parallel monolayer, spinner, and RWV MSCs after 7 days of culture. We confirmed that all MSC markers were expressed by a high percentage of monolayer MSCs and lacked expression of the hematopoietic markers CD34 and CD45. However, MSCs from spinner flask and RWV cultures showed a decrease in the expression levels of all markers, with a particularly large decrease in the expression of CD29, CD105, CD166, and HLA class I (Table 1). These changes in surface antigen expression profile observed in dynamic 3D culture were reversible and the expression levels returned when the cells were replated onto tissue culture plastic and grown as two-dimensional (2D) monolayers (data not shown). Analysis of the forward scatter properties of monolayer and dynamic 3D MSCs also revealed that after 3 and 7 days of culture the populations from dynamic 3D culture had decreased in size when compared with monolayer MSCs. The distribution of cell sizes also showed a more homogeneous distribution than the large spread of the monolayer cells (Fig. 3C). There were no changes in the side-scatter properties.
The expression level of cell surface antigens was determined after 7 days of monolayer, spinner flask, and RWV cultures. Percentages shown are the proportion of positively stained cells relative to an isotype control.
RWV, rotating wall vessel; HLA, human leukocyte antigen.
Osteogenic differentiation is enhanced in dynamic 3D MSC cultures
As 3D MSCs lacked expression of some previously described MSC surface antigens, 24 it was important to determine if this also reflected a change in their stem/progenitor cell characteristics and differentiation potential. Monolayer, spinner, and RWV MSCs were cultured in the presence of osteogenic supplements for 21 days, with samples taken after 7, 14, and 21 days for analysis by histological staining and qRT-PCR.
We identified alkaline phosphatase staining in monolayers from 14 days onward, but matrix mineralization was only observed after 21 days of differentiation and at low levels (Fig. 4A). Alkaline phosphatase was detected in RWV MSCs at days 14 and 21 and in spinner MSCs it was present after 7 days of differentiation. Both spinner and RWV MSCs showed discrete areas of von Kossa-positive mineralization from day 14 onward.

Osteogenic and adipogenic differentiation are enhanced in dynamic 3D MSC cultures. Osteogenic or adipogenic supplements were added to MSCs for 21 days in monolayer, spinner, and RWV cultures. Samples were taken at days 7, 14, and 21 to establish the extent of differentiation by histological staining and qRT-PCR analysis of the relative expression levels of marker genes. (
Determination of relative gene expression levels by qRT-PCR demonstrated a decreased level of alkaline phosphatase in spinner and RWV MSCs, compared with monolayers, and no significant changes in Cbfa-1 expression. However, there were increased levels of later osteogenic markers, osteocalcin and osteopontin, in both the dynamic 3D cultures. Osteocalcin expression decreased in monolayer MSCs but was significantly increased in spinner and RWV MSCs after 7 and 14 days of differentiation and in spinner MSCs on day 21. Osteopontin expression also decreased in monolayer MSCs but increased in dynamic 3D MSC cultures. At all time points, levels of osteopontin were significantly higher in both the dynamic 3D cultures than monolayer MSCs (Fig. 4C).
Adipogenic differentiation is enhanced in dynamic 3D MSC cultures
The level of adipogenic differentiation was compared between monolayer and dynamic 3D MSCs after 7, 14, and 21 days of culture with the addition of adipogenic supplements. Oil Red O staining showed lipid droplet accumulation on day 7 in spinner and RWV MSCs. These showed an increase in both size and number on days 14 and 21. Lipid droplets were distinguishable only in monolayer cultures on day 21 (Fig. 4B).
qRT-PCR analysis of adipogenic marker gene expression levels showed increased expression of PPARγ, C/EBPα, LPL, and aP2 in all cultures after the induction of adipogenesis (Fig. 4D). The expression level of all four markers was significantly increased in RWV MSCs compared with monolayer MSCs. Spinner MSCs also showed increased expression of PPARγ and aP2 compared with monolayers on day 7 and increased expression of C/EBPα on days 7 and 14.
Expression profiling of dynamic 3D MSCs
Expression profiling was performed to further examine the differences between monolayer and dynamic 3D MSCs. As MSCs from both of the dynamic cultures showed equivalent viability, surface marker expression, and differentiation potential, spinner flask spheroids were chosen as a representative model to be used to examine gene expression in dynamic 3D MSCs. Initial correlation analysis, in which a value of 1 indicates identical gene expression levels in two different MSC cultures, showed an average correlation of 0.947 within monolayer samples, 0.962 within spinner samples, and 0.870 when comparing monolayer and spinner samples, indicating a change in gene expression profile between monolayer and dynamic 3D MSCs. t-Tests identified 710 genes that were differentially expressed at a significance level of 1% and fold change of ≥2. Of these, 277 gene transcripts were downregulated (Table 2) and 433 gene transcripts were upregulated (Table 3) in dynamic 3D MSCs compared with monolayers. To validate these results, representative genes covering the whole range of significantly up- and downregulated transcripts were selected and qRT-PCR performed to confirm their expression levels in the two culture systems. This was performed using MSCs from a donor independent to those used for the microarray analysis. This confirmed the changes in expression determined by the array analysis giving confidence in the accuracy of the array data as a whole (Fig. 5).
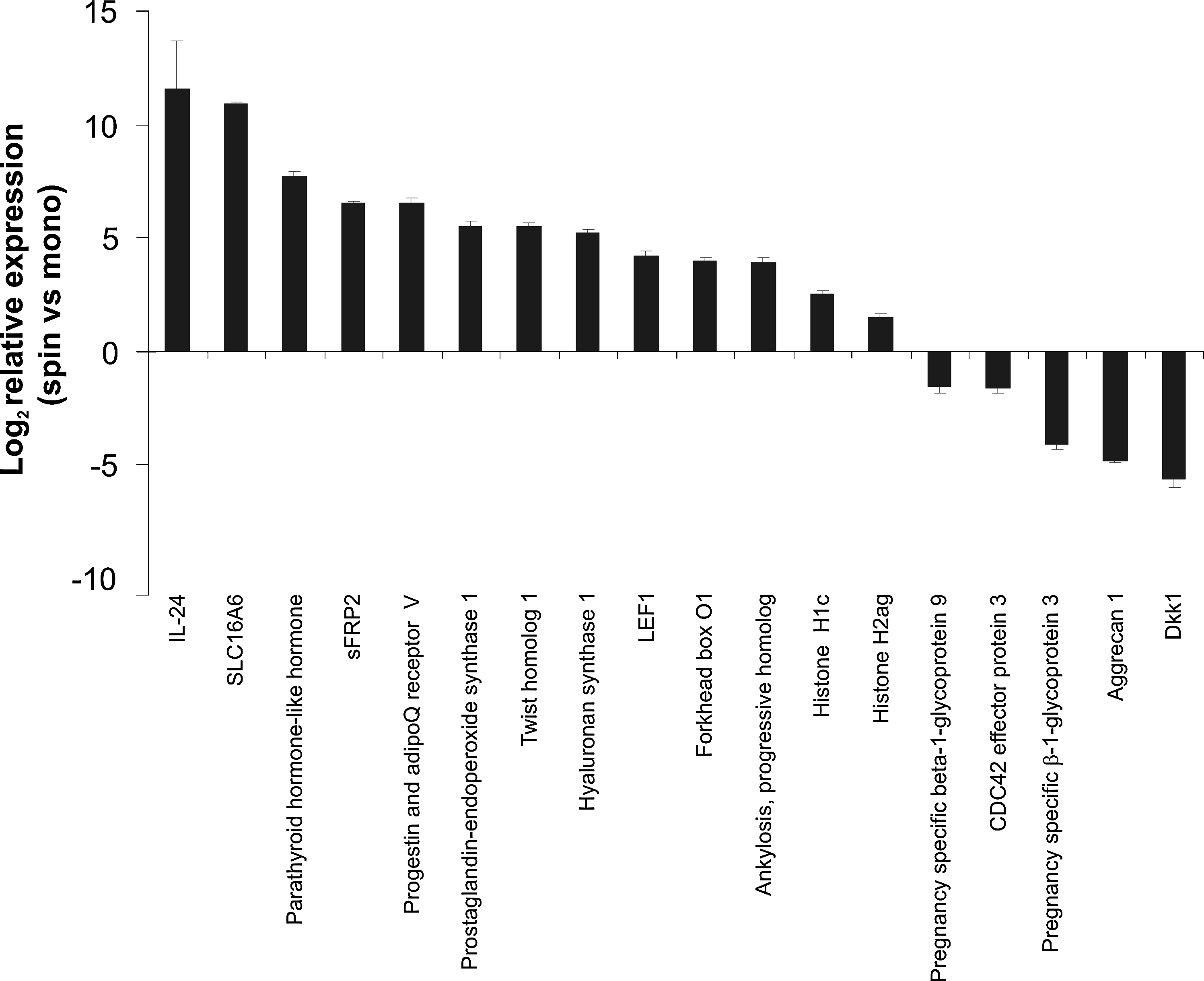
qRT-PCR validation of the microarray data. qRT-PCR was performed to determine the expression level of a number of the genes determined to be significantly up- or downregulated in dynamic 3D MSCs compared with monolayer MSCs. These data were obtained using MSCs from a donor independent to those used for the microarray analysis. Data are shown as mean expression, relative to monolayer MSCs (± standard error of the mean). IL24, interleukin 24; sFRP2, secreted Frizzled-related protein 2; SLC, solute carrier protein; LEF1, lymphoid enhancer-binding factor 1.
The table shows the 50 genes most downregulated in dynamic three-dimensional mesenchymal stem cells (MSCs) when compared with monolayer MSCs. The table includes details of the Affymetrix probe, gene name, and symbol as well as the average fold change across all three donors and the p-value for this change.
The table shows the 50 most upregulated genes in dynamic three-dimensional mesenchymal stem cells (MSCs) when compared with monolayer MSCs. The table includes details of the Affymetrix probe, gene name, and symbol as well as the average fold change across all three donors and the p-value for this change.
The differentially regulated gene transcripts were classified according to function using GO terms, with annotations successfully assigned from the current GO database to 225 of the 227 downregulated transcripts and 261 of the 433 upregulated gene transcripts. The downregulated transcripts demonstrated significant enrichment in the categories of biological adhesion, developmental processes, and multiorganism processes as well as in motor activity, structural molecule activity, enzyme regulator activity, and ECM, whereas the upregulated transcripts showed enrichment in a broader range of categories which, like the downregulated transcripts, included biological adhesion and developmental processes and also antioxidant activity, response to stimuli, and signal transducer activity.
IL24 in medium conditioned by dynamic 3D MSCs impairs the viability of cancer cells
To investigate the functional effects of the gene expression changes induced by the altered culture conditions and with a particular focus on the fact that there were changes to many signaling factors, we devised a simple screen to look for differences induced by factors synthesized by MSCs from each of the culture environments. Culture medium conditioned by monolayer or spinner flask MSCs was used to treat both MSCs and other cells from the bone marrow microenvironment and viability was determined by MTT assay. The results showed no difference in the viability of primary MSCs, C3H10T1/2, or human umbilical vein endothelial cells cultured in 2D and 3D conditioned medium. However, the viability of the human osteosarcoma cell line Saos-2 was significantly decreased in cells from 3D versus 2D conditoned media (Fig. 6A). The apparent difference in the response of cancerous cells compared with noncancer cell types was investigated further by determining the viability of a number of cancer and noncancer cell lines in 2D and 3D conditioned media. These results showed no difference in viability of the normal immortalized PNT-1a and PNT2-C2 prostate cell lines but a decrease in the viability of the cancerous PC3, VCAP, LNCAP, and DU145 cell lines when cultured in medium conditioned by dynamic 3D MSCs (Fig. 6B). Referring back to the array data it was hypothesized that this difference may be attributable to the 75-fold upregulation of IL24 in the dynamic 3D MSC cultures because IL24 has been determined to impair the viability of cancer cells without affecting the viability of normal cells.25–27 To confirm this, the viability of LNCAP prostate cancer and PNT-1a normal prostate cells was determined after 4 days of culture with 2D and 3D conditioned media with the addition of an antibody that blocks the action of IL24. Interestingly, there were no significant differences in the viability of PNT-1a cells in any of the treatments but the reduction in viability of LNCAP cells cultured in 3D conditioned medium was reversed, in a dose-dependent manner, by the addition of the IL24 blocking antibody (Fig. 6C).
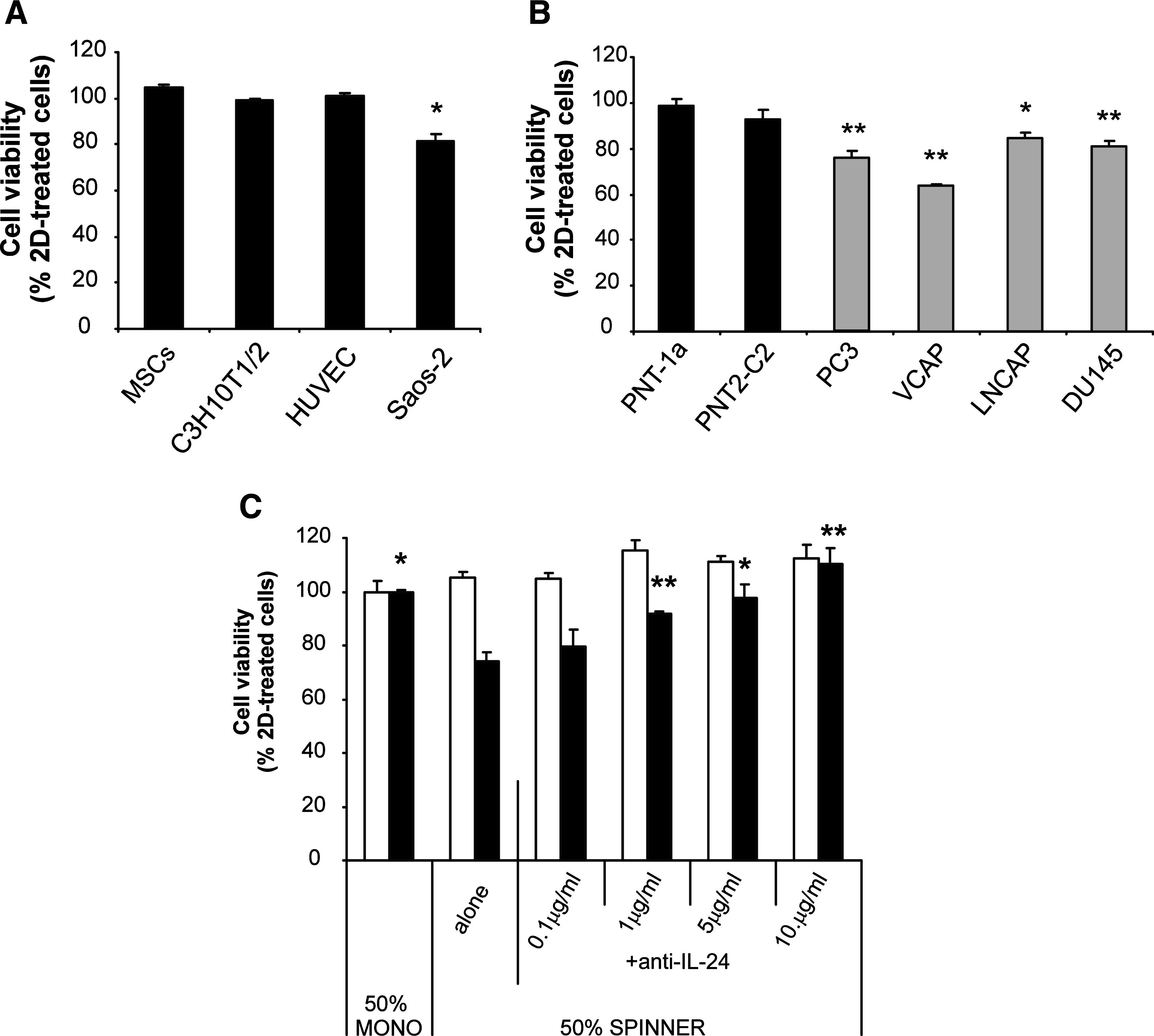
IL24 synthesized by dynamic 3D MSCs specifically reduces the viability of cancer cells. Conditioned medium from monolayer (2D) and spinner flask (3D) MSC cultures was used to treat a number of cell types for 4 days before determination of the cell viability by MTT assay. All graphs show cell viability in 50% 3D conditioned medium relative to that of a parallel culture treated with 50% 2D conditioned medium. Bars represent the mean percentage (± standard error of the mean), n = 3, *p < 0.05, **p < 0.01. (
Discussion
MSCs lose many of their beneficial properties after prolonged periods of in vitro culture using standard 2D techniques. We developed and characterized dynamic 3D techniques for in vitro MSC culture using spinner flasks and the National Aeronautics and Space Administration RWV, which may more accurately model in vivo MSC biology. A comparison between these dynamic 3D and monolayer MSCs identified many changes in the cells, which confirms the hypothesis that the dynamic 3D environment has an influence on MSC properties. Our data suggest that dynamic 3D techniques may be beneficial for MSC maintenance and differentiation but also provide insights into novel ways by which such MSCs may be used for therapeutic applications.
We demonstrated that MSCs aggregate to form spheroids when cultured in dynamic 3D conditions. However, MSCs placed directly into dynamic conditions as a single-cell suspension did not form aggregates and were not viable. This suggests that the initial period of static high-density culture was vital to allow the cells to make cell–cell contacts and thus overcome anoikis mechanisms. 28 Under the described conditions, the cell viability and turnover on day 7 remained equivalent to that of monolayer MSCs, confirming that the dynamic 3D techniques are a viable alternative to monolayer culture. Alternative techniques to increase cell numbers in 3D growth could be achieved through repeated rounds of disaggregation followed by reaggregation into 3D spheroids.
There was no evidence of cell death in the center of the spheroids, as is often seen with tumor cells. 29 This is likely to be due to the small size of the spheroids, particularly those from RWV culture, and it suggests that the spheroid diameter allows sufficient diffusion of oxygen and nutrients into the spheroid core. The dynamic conditions may also help with this, through the movement of nutrients to and waste products away from the spheroids in culture which has previously been noted to aid the viability of cells in the center of other tissue constructs cultured in vitro.30,31
Our findings demonstrated that dynamic 3D MSCs maintain their multipotent differentiation potential. We focused on osteogenic and adipogenic differentiation, as chondrogenesis is known to be maintained in 3D micromass cultures. We demonstrated enhanced differentiation in both spinner and RWV spheroids when compared with monolayer MSCs. This was most clearly displayed by adipogenic cultures with increased Oil Red O staining and expression of adipogenic markers in dynamic 3D MSCs compared with monolayers. The osteogenic cultures showed no detectable increase in the transcript levels of the early markers alkaline phosphatase and Cbfa-1; however, cytochemical analysis did detect increased alkaline phosphatase activity in dynamic 3D MSCs. This result highlights the fact that there can sometimes be discrepancies between transcript levels and the amount of functional protein produced, but together with the increased expression of later osteogenic markers osteocalcin and osteopontin and increased von Kossa staining, the overall results indicate enhanced osteogenic differentiation of the MSCs in dynamic 3D conditions.
We attribute the enhanced differentiation to a combination of factors from both the 3D and dynamic aspects of the culture conditions. Our 3D spheroids provided increased cell–cell contacts, which have been shown to improve differentiation. 32 The maintenance of the spheroids in a dynamic suspension culture also meant that the MSCs adhered only to other cells and their natural ECM and it has been shown that providing MSCs with a more natural matrix to adhere improves their ability to differentiate.33–35 The difference in tension between MSCs adhering to a rigid plastic surface and the more elastic ECM may also have had an effect, as matrix elasticity can direct stem cell fate. 36 There is also evidence to suggest a link between cell shape and lineage commitment, with flattened MSCs more able to become osteoblasts and rounded cells more able to differentiate into adipocytes. 37 This may help to explain why the cells within our spheroids were able to differentiate so efficiently along the adipogenic lineage. The enhanced differentiation in response to both osteogenic and adipogenic factors is also consistent with data from Wang et al., 38 who using a static MSC spheroid model showed also a decrease in CD90 expression in 3D MSCs and suggested that the loss of expression may indicate that the MSCs are primed and ready to differentiate upon the addition of appropriate signals. Further work would be needed to confirm such a mechanism, but if this was the case it could also apply to our dynamic spheroid cultures.
Dynamic culture conditions are often used to seed cells onto constructs 18 and also to produce an environment with mechanical forces. MSCs cultured on scaffolds in dynamic conditions have demonstrated improved osteogenesis compared with static cultures, a result that has been attributed to the increased shear stress. 16 Spinner flask cultures have high level of shear stress, whereas the RWV culture environment has very low levels. We found enhanced osteogenesis of MSCs in both of these bioreactors compared with monolayer MSCs but this may be a result of changes in the 3D environment of the spheroids as well as the dynamic conditions. The RWV has also previously been used to promote differentiation of MSCs, although these studies rely on a scaffold or carrier rather than spheroid system. These studies have primarily shown an increase in adipogenesis and decrease in osteogenesis 39 although results differ. Our data showed both enhanced osteogenesis and adipogenesis in the RWV. Again, as suggested by work using static MSC spheroids which also show enhanced differentiation along both lineages, 38 this is likely to be due to factors from the 3D culture as well as the dynamic conditions. However, it has been suggested that RWV culture can increase osteogenesis using small cellular constructs, consistent with the small size of MSC spheroids in the RWV. 21 Adipogenesis was increased further in the RWV compared with spinner cultures and 2D cultures, supporting evidence that the RWV is an improved environment for adipogenic differentiation of MSCs.
Although we demonstrated that 3D MSCs maintained both osteogenic and adipogenic differentiation potential, we also showed decreased expression of some cell surface antigens, often used as MSC markers. 24 We do not believe that these changes indicate a loss of stemness in the dynamic 3D cultures as the microarray data did not show changes in the expression of genes associated with differentiation in MSCs cultured in basal medium. CD29, CD105, and CD166 are involved in cell–cell and cell–ECM interactions 40 ; therefore, we believe that their loss of expression reflects the change between attachment of MSCs to a plastic surface and the 3D environment in which the MSCs make contacts with other cells and the ECM. The expression profile of MSCs is defined as HLA class I positive and class II negative. 24 The decreased HLA class I in dynamic 3D MSCs is closer to that seen in fetal MSCs 41 or embryonic stem cells (ESCs) which are characterized by weak surface expression of HLA class I which increases upon differentiation. 32 The dynamic 3D MSCs also decreased in size, possibly because of the fact that the cells were compacting into a spheroid rather than spreading out on plastic, and resulted in a cellular phenotype with a high nuclear:cytoplasmic ratio, typical of ESCs or cells with a primitive phenotype. This loss of HLA class I in dynamic 3D MSCs as well as the decreased cell size could reflect a more primitive MSC phenotype and may prove a major therapeutic benefit, providing a method to generate MSCs with reduced immunogenicity which will be more useful for transplantation and cell-based therapies.
Our observations led us to a broader analysis of the gene expression repertoire of 3D MSCs versus 2D MSCs and we demonstrated by microarray analysis many changes between monolayer and dynamic 3D MSCs. Previous studies have determined the expression profiles for MSCs from different tissue sources, 42 throughout differentiation43,44 or in response to the addition of soluble signaling factors,45,46 but this is the first time that a change in gene expression has been determined when MSCs are subjected to an altered 3D and dynamic environment.
The GO enrichment analysis allowed the identification of categories that were enriched in the up- and downregulated genes. The enrichment of transcripts classified under biological adhesion, structural molecule activity, and ECM highlighted changes to the cytoskeleton. This included the downregulation of a number of keratins and keratin-associated proteins, smooth muscle actin, tropomyosin 1, and several myosins and corresponds well with the fact that the MSCs changed in morphology when cultured as 3D spheroids. The ECM and factors involved in the synthesis and turnover of the ECM were also highlighted with a decrease in the expression of various factors that comprise or are involved in remodeling of the ECM, for example, the downregulation of aggrecan, collagen XI and XII, and lysyl oxidase and upregulation of spondin 1 and hyaluronan synthase. Some of these changes may reflect an adjustment of the cells to the mechanical environment of the spinner flask culture, as matrix proteins are often differentially regulated in response to stress and strain.47,48 These changes are likely to have implications for the function of the cells, as ECM not only provides structure and a substrate for the cells to adhere but also to regulate the availability of various growth factors and cytokines which in turn has implications for cell survival and differentiation. 49
Other differentially expressed genes also indicated adaptation to the 3D culture conditions. The striking 1000-fold upregulation of the monocarboxylate transporter SLC16A6 was just one change in the expression of a number of solute carrier protein (SLC) family members. Expression of SLC16 family members has been linked to hypoxia 50 and is increased in MSCs cultured under conditions of low oxygen tension. 51 This change is likely to be because of reduced oxygen availability within the compact 3D spheroids but may be a positive factor because hypoxic conditions are thought to be present within the MSC niche 52 and have been demonstrated to maintain MSCs in the undifferentiated state. 53 The increase in FoxO1 expression also supports an apparent change in metabolic activity. FoxO1 expression is regulated by nutrient availability and oxidative stress 54 and has previously been demonstrated to increase under dynamic 3D culture conditions in ESCs. 55
The altered expression of a number of transcripts classified under developmental process, including upregulation of chemokine (CXC motif ) receptor 4, IL24, parathyroid hormone-like hormone, and secreted Frizzled-related protein 2 and downregulation of connective tissue growth factor, Chordin-like 1, Noggin, and Dkk1 lead us to hypothesize that the altered expression of many secreted signaling factors by 3D MSCs may influence the behavior of 2D MSCs as well as a range of other cell types. We found no effect of 3D MSC-conditioned medium on MSC viability. However, we found that 3D conditioned medium decreased the viability of all cancer cell lines tested, but not that of normal immortalized cell lines, and we hypothesized that this was due to the upregulation of IL24 revealed by our microarray analysis. IL24 is a member of the IL10 family of cytokines and has received growing interest because of reports that it can specifically impair the viability of cancer cells while leaving normal cells unaffected.25–27 From our data we propose that MSCs may be a novel source of IL24 for future therapies. Such a method could have an advantage over current approaches that rely on adenoviral-mediated ectopic expression of IL24 to exert an effect. 25 When coupled with the fact that our dynamic 3D MSCs also express increased levels of chemokine (CXC motif ) receptor 4, MSCs cultured under dynamic 3D conditions could also provide a new prospect for a cell-based therapy in which the MSCs were targeted to diseased areas with high SDF1 expression where the increased levels of IL24 could exert an effect. This proposition, however, would require further substantial validation, including in vivo analysis of 3D MSC phenotype stability. However, our findings do support previous work showing that MSC populations can home to tumors in vivo with potent growth-inhibiting effects.56,57
We have used dynamic 3D methods for in vitro MSC culture and shown that MSCs cultured in this way have different and, in many ways, improved properties when compared with those in monolayer culture. Ex vivo analysis of stem cell populations is an essential research tool, but it is prone to culture-induced artifacts. There is growing evidence that 2D cell monolayers lack the required sophistication, particularly considering the growing knowledge of 3D stem cell niche.
Footnotes
Acknowledgments
The study was funded by the BBSRC and Smith and Nephew. The authors thank Harrogate District Hospital for providing bone samples, Prof. Normal Maitland and Dr. Anne Collins (University of York, UK) for providing the prostate cancer cell lines, and Catherine McGrath for expert technical assistance in the preparation of this work.
Disclosure Statement
The authors declare no competing interests.