
Abstract
Transgenic mice with a Col1a1-promoter-driven transgene pOBCol2.3GFP were previously developed to visually identify mature osteoblasts through fluorescent expression. Our goal was to determine if this technology could be used to nondestructively evaluate the in vitro differentiation of osteoprogenitor cells on biomaterials such as biomimetic carbonated hydroxyapatite (cHA). Primary osteoprogenitor cells were harvested from calvaria of neonatal Col2.3GFP transgenic mice and cultured on cHA and a tissue culture polystyrene (TCPS) control. The distribution of intensities and area percentage of green fluorescent protein (GFP)–positive cells were quantified using fluorimetry and image analysis of fluorescent microscopy. At 14 days, an increased area and higher mean intensity of GFP-positive cells was observed on cHA as compared to TCPS, indicating more rapid differentiation on cHA. Notably, there were large continuous regions of GFP-positive osteoblasts on cHA, in contrast to the sparse, nodules of osteoblasts on TCPS, implying that cHA provides an osteogenic cue to cells. Xylenol orange staining was capable of distinguishing osteoblast-initiated mineral from the cHA substrate. With this method the unique pattern of osteoblast differentiation on cHA was clearly observed for the first time. Importantly, the generalized method can be used for rapid, high-throughput, nondestructive screening of biomaterials intended to enhance osteogenic differentiation.
Introduction
The use of ubiquitous fluorescent protein expression to observe cells and their structures is a widely accepted technique. 1 The method has been applied in tissue engineering to observe and quantify the number of genetically modified rat fibroblastic cells within a hydrogel and on a titanium mesh. 2 The response of continually fluorescent mouse mesenchymal cells to three different hydrogels in vivo 3 and in a calvarial bone repair model 4 has been reported. However, all previous investigations of fluorescent cells in biomaterials have used ubiquitous markers, meaning that all of the cells fluoresced all of the time. In contrast, the cells used in the present study only fluoresce when the progenitor cells differentiate to mature bone cells—before that they are not fluorescent. 5 Such cells are available from transgenic mice specifically developed for the purpose of monitoring osteoblast lineage status.5,6 They express green fluorescent protein (GFP) only upon becoming a mature osteoblast due to the Col1a1-promoter-driven transgene pOBCol2.3GFP.5,6 GFP expression within pOBCol2.3GFP mice has been correlated to osteogenic marker genes showing that the GFP expression level directly and reliably identifies late-stage osteoblasts capable of secreting mineralized matrix.5–7 Related transgenic mice have been developed for other stages of osteoblast differentiation and include (1) α-smooth muscle actin–GFP reporter mice with cells that fluoresce during the early progenitor stage, 8 (2) pOBCol3.6GFP mice with cells that fluoresce during the preosteoblast stage (alkaline phosphatase expression stage), 5 and (3) osteocalcin-GFP mice for even later stage osteoblasts and osteocytes than Col2.3GFP. 9 Other cell types in these mice do not express the osteoblast-associated GFP. Osteoprogenitor cells from pOBCol2.3GFP transgenic mice can provide a reliable approach to continuously monitor osteoblast differentiation in real time either in vivo or in vitro. In the present study, we tested the hypothesis that the status of osteoblasts differentiating on biomimetic calcium phosphates (CaP) could be evaluated qualitatively and quantitatively during in vitro cultures through the expression of stage-dependent fluorescent markers.
CaP have been used extensively for bone repair because of their osteoconductive/osteoinductive and degradation characteristics.10–13 These same properties make CaP effective as matrices or constituents of scaffolds for cell-based tissue engineering of bone.14–16 Accordingly, there has been considerable study of the in vitro interactions between mineral-forming cells and CaP.13,17–20 Typical experiments of osteogenic differentiation on CaP involve manipulation of the surface composition or topography and monitoring of cell attachment, proliferation, and differentiation. Sintered hydroxyapatite (HA) or biomimetic carbonated HA (cHA) is the particular form of CaP most studied.13,17–20 From these studies, discernable trends regarding the influence of HA have been observed. Initial cell attachment is lower on cHA and HA compared to tissue culture polystyrene (TCPS), but the differences later subside. 19 Depending on the time point proliferation also may be inhibited on HA compared to TCPS.19,21 However, differentiation appears to be accelerated by HA or cHA. Alkaline phosphatase and messenger RNA 22 and osteocalcin 17 are increased on HA, and matrix production per cell appears higher. 19 HA has been shown to increase differentiation of stromal cells toward an osteogenic fate.23,24 With mouse embryonic stem cells, HA affects differentiation and can induce osteo-specific genes. 25 The exact mechanism of action remains unknown and will not be solved unless cell-specific studies are undertaken.
Cell-specific studies on CaP biomaterials are currently hindered because the nontransparent nature of the scaffolds makes it difficult to study cell morphology and distinguish mineral deposition from the original scaffold. The autofluorescence of CaP, particularly HA or cHA, and the high binding affinity of the stains for mineralized matrix to CaP also complicate analyses of osteogenesis on CaP. Xylenol orange (XO) has recently been demonstrated as a means of staining the mineralized matrix of vital osteoblast cultures grown on TPCS. 26 We therefore investigated the suitability of this stain to distinguish mineralized matrix deposited by cells from a mineral substrate: cHA.
Here we describe the use of lineage-specific promoter-driven GFP in combination with vital XO staining to monitor and quantify mouse calvarial progenitor cell differentiation during an in vitro mineralization assay on cHA and TCPS. Use of this method allowed, for the first time, visual observations of osteogenic responses to cHA at the individual cell level without requiring the termination of the culture. Importantly, the generalized method can be used for rapid, high-throughput, nondestructive screening of biomaterials intended to enhance osteogenic differentiation.
Materials and Methods
Preparation of the cHA disk inserts
A cHA coating was applied to 34.5-mm-diameter disks cut from 100-mm standard tissue culture–treated plastic dishes (BD Falcon/Fisher) following a recently described procedure. 27 Briefly, a two-step SBFx5 biomimetic cHA coating protocol that had been developed for metal substrates28,29 was modified to coat the TCPS disks. To obtain an adherent coating on TCPS, sandblasting of the substrate, a short postcoating sonication step, and dehydration in a series of graded ethanol between and after each precipitation step were included. The chemical and physical properties of the biomimetic coating were characterized by several techniques (X-ray diffraction, Fourier transform infrared spectroscopy, profilometry, and energy dispersive X-ray spectroscopy) and found to be 100% low crystalline carbonated apatite. 27 The coating is approximately 10 μm thick with a sharp, nano-scale crystalline topography (Fig. 1A). The cHA-coated disks were used as cell culture well inserts and were sized to fit snugly into six-well tissue culture plates.

(
Harvesting the GFP cells
Primary mouse calvarial osteoblast progenitor cells (mCOBs) were harvested from the calvaria of 5–8-day-old neonatal transgenic mice carrying a GFP gene driven by a 2.3-kb fragment of the type 1 collagen promoter, following a published protocol with modifications. 5 Briefly, after removal of sutures, calvaria were subjected to four sequential 15-min digestions in an enzyme mixture containing 0.25% trypsin and ethylenediaminetetraacetic acid (Gibco BRL) and 2 U/mL collagenase P (Boehringer Mannheim) at 37°C on a rocking platform. Cell fractions 2–4 were collected and enzyme activity was stopped by the addition of an equal volume of the culture medium containing Dulbecco's modified Eagle's medium (#11885; Gibco), 10% fetal bovine serum, 100 U/mL of penicillin, 100 μg/mL of streptomycin, and 0.1 mM nonessential amino acid (Gibco BRL). The fractions were pooled, centrifuged, resuspended in the culture medium, and filtered through a 70-μm cell strainer.
Culturing the cells on the cHA inserts and TCPS controls
The cHA-coated disks were sterilized by exposure to UV light for 1 h, and then immersed in Dulbecco's modified Eagle's medium with 10% serum for 2 h at 37°C before seeding the cells. Cells were seeded at a density of 1.5 × 104/cm2 on both the control tissue-culture-treated six-well plates (TCPS; BD Falcon/Fisher) and the cHA-coated disk inserts placed in non-tissue-culture-treated six-well plates (BD Falcon/Fisher). After 4 and 48 h, cells on the cHA inserts and TCPS controls were examined with an inverted fluorescence microscope (Eclipse TE 300; Nikon, Inc.) to observe the cell morphology. At these early time points, the cells were not yet GFP positive, and thus a Calcein-AM dye (Calcein acetoxy-methyl ester BD Bioscience) was used according to the manufacturer's specifications to more clearly observe the cells. More complete cell attachment and proliferation studies on cHA and TCPS have been completed previously with these cells, as well as the MC3T3-E1 cell line.
27
In the present study, the culture medium (described above) was changed every other day for a week until the cells became confluent on the tissue culture plastic controls, after which a differentiation medium consisting of α-minimal essential medium, 10% fetal bovine serum, 100 U/mL penicillin, 100 μg/mL streptomycin, 50 μg/mL
The distribution of all of the cells, including non-GFP-positive cells, on the materials was observed using DAPI nuclear staining (Molecular Probes). It was not possible to quantify cell number by image analysis using DAPI staining due to multicell layers and cell overlap at the later time points, particularly on the TCPS in which the osteoblasts formed the typical three-dimensional osteoblast nodules.30,31 Therefore, at 3, 7, 14, and 21 days, relative total cell number was determined by measuring DNA content for three wells each of cHA and TCPS. Cell extracts were prepared by applying 1 mL TRIzol reagent (Invitrogen) to each culture well at room temperature, allowing 30 min for extraction. Mechanical scraping was needed to completely release some cells in the 14- and 21-day cultures. The double-stranded DNA content in the cell extracts was measured with a fluorescent nucleic acid assay (PicoGreen; Molecular Probes) according to the manufacturer's protocol.
Imaging and quantifying GFP with fluorescence microscopy
GFP expression of the cells on the cHA and TCPS control surfaces was quantified by analyzing images from fluorescence microscopy at 3, 7, 14, and 21 days. To observe large areas of the cultures a computerized stereo microscope (Zeiss SteREO Lumar V12; Carl Zeiss) was used with a Topaz (yellow fluorescent protein) filter (Chroma Technology). The microscope is equipped with a motorized X-Y-Z stage, motorized fluorescence cube, and color digital camera (AxioCam; Carl Zeiss) controlled by a user-defined computation program. The exposure time was optimized during pilot studies so that images of day 21 cultures would not be overexposed, which would prevent accurate quantification due to loss of data at the highest grayscale levels. These settings were then used for all time points and for both cHA and TCPS in the full study. A series of 12 × 4 adjacent images with 15% overlap was taken of each well. Each series of images was concatenated into a single image that spanned 29 mm of the midsection of the 35-mm well diameter and covered 22.5% of the 9.6 cm2 area of the well. Initially, images were captured for the entire well, but to reduce the amount of time the plate was out of the incubator and the image analysis was optimized to collection of 48 separate images, which gave equivalent values to whole well analysis. The fluorescence intensity of each pixel in the concatenated image was captured at each time point. These data were in relative fluorescence units and had possible gray-scale values of 0–255.
Quantification of the area percentage of GFP-positive cells and mean GFP intensity was completed using basic image analysis software (ImageJ; National Institutes of Health). Background fluorescence was eliminated from the area analysis by enlarging the image and selecting a threshold level that excluded the non-GFP background and resulted in a visual match between the thresholded image and the GFP-positive areas in the fluorescent microscopy image. The very bright GFP fluorescence relative to the autofluorescence of the cHA and the relative on/off nature of the Col2.3GFP expression made this thresholding procedure straightforward and a reproducible process that was repeated by several users in the laboratory. A threshold level of 14 out of the possible 255 was used for all images at all time points. The mean intensity of the GFP-positive pixels within the thresholded images was computed using ImageJ. Mean intensity values were also calculated with an alternative background subtraction procedure in which values of mean intensity from nonthresholded day 3 images were subtracted from the nonthresholded day 14 and 21 images and found to be identical to the contour-matching approach. Area and intensity values were normalized to total cell number determined by DNA content analysis.
Quantifying GFP with fluorimetry
In addition to imaging and quantification with fluorescence microscopy, the mean GFP fluorescence intensity of each well was also measured with a multidetection monochrometer microplate reader (Safire II; Tecan), with 490 nm excitation, 510 nm emission, and 10 nm bandwidth. The entire area of each well was read at a scan density of 9 × 9 regions and a gain of 120. Background intensities were measured using separate six-well plates, with and without cHA, with cultures of wild-type, nonfluorescent mCOBs; harvested; and cultured without β-glycerophosphate, thereby preventing mineralization. The average background intensity for cHA under the GFP filter set was 1.3× TCPS at all time points, while the GFP differences were on the order of 7×. The control background measurements for each substrate were subtracted from their GFP measurements. This background subtraction technique eliminates any contribution from the indicator dye contained in the medium as well as any autofluorescence of the cHA. The autofluorescence of the cHA under the GFP filter set at the exposure times used in this study was essentially nondetectable as evidenced by the unadjusted dark background of the images.
Staining, imaging, and quantifying mineralized nodules
XO, a nontoxic calcium-chealating fluorochrome reagent that labels newly calcified tissues, 26 was used to nondestructively observe mineralized modules in the cell cultures at 14 and 21 days. XO powder (Sigma–Aldrich) was dissolved in distilled water and filtered to make a concentrated 20 mM stock and stored at 4°C. XO was added to the culture medium at a final concentration of 20 μM at least 12 h before imaging. This concentration of XO has been shown previously to label mineralized osteoblast nodules on TCPS similarly to von Kossa staining without any detectable loss of cell viability. 26 The XO-containing medium was exchanged for a fresh medium before imaging to avoid the nonspecific fluorescent background from unbound XO. Images of XO samples were taken with the Zeiss SteREO Lumar V12 computerized stereo microscope used with a custom Cherry filter (Exciter ET577/20, Emitter ET640/40; Chroma Technology). To observe more closely the mineralization and the cHA substrate features at higher magnification, images were also obtained with a Nikon Eclipse TE 300 microscope (Nikon, Inc.) equipped with fluorescent filters for GFP, XO, and DAPI.
The fluorescence intensity of the XO-stained mineral throughout the entire well was quantified with a fluorimeter (Safire II; Tecan) with 546 nm excitation, 580 nm emission, 10 nm bandwidth, 9 × 9 scan density, and a gain of 180. To correct for background fluorescence from the biomaterials and medium, controls of wild-type mCOBs were cultured on each surface in a β-glycerophosphate-free medium to prevent mineralization of the extracellular matrix and read at the same time points as the Col2.3GFP cultures. These controls were particularly important for the XO analysis, because like the mineralized matrix, the cHA also absorbs XO. The fluorometric control readings were subtracted from the experimental values.
Statistical analysis
t-Test statistical analysis at a significance level of p < 0.05 was used to identify differences in cell response to the cHA and TCPS surfaces at each time point. Data are presented as the mean ± SD for three replicates. The entire mineralization experiment was repeated five times to ensure reproducibility.
Results
mCOB cell morphology at 4 and 48 h
Representative cell morphologies on the TCPS and cHA surfaces at 4 and 48 h are shown in Figure 1B. After 48 h of incubation the mCOB cells on the TCPS spread more fully than those on cHA. Cells on the TCPS formed broad lamellipodia, and the extended protrusions are evidence of migration activity, which is not seen on the cHA. In contrast, after 48 h the cells on the cHA were slender with elongated processes.
GFP expression
Concatenated fluorescence micrographs of the GFP-positive cells on the cHA and TCPS surfaces at 14 and 21 days are shown in Figure 2A. At 3 and 7 days, GFP expression was not visible using the exposure times selected for these studies (images not shown). After 14 days of culture, the GFP-positive cells on TCPS were sparsely distributed across the culture well in multicell layer nodules, while larger areas were GFP positive on the cHA (Fig. 2A). At 21 days, GFP expression was increased on TCPS, and considerably more of both culture surfaces were covered by GFP-positive cells (Fig. 2A).
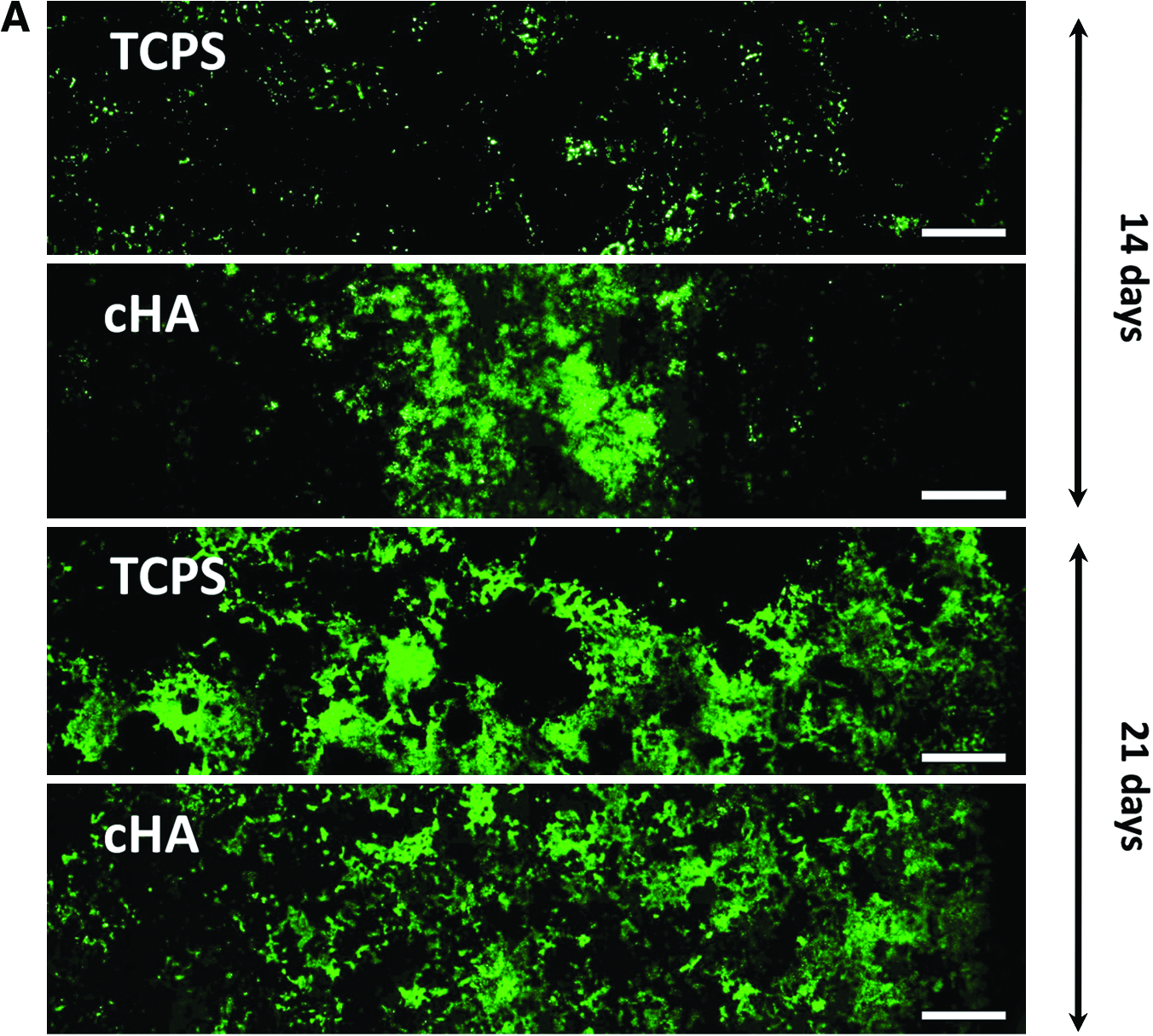
(
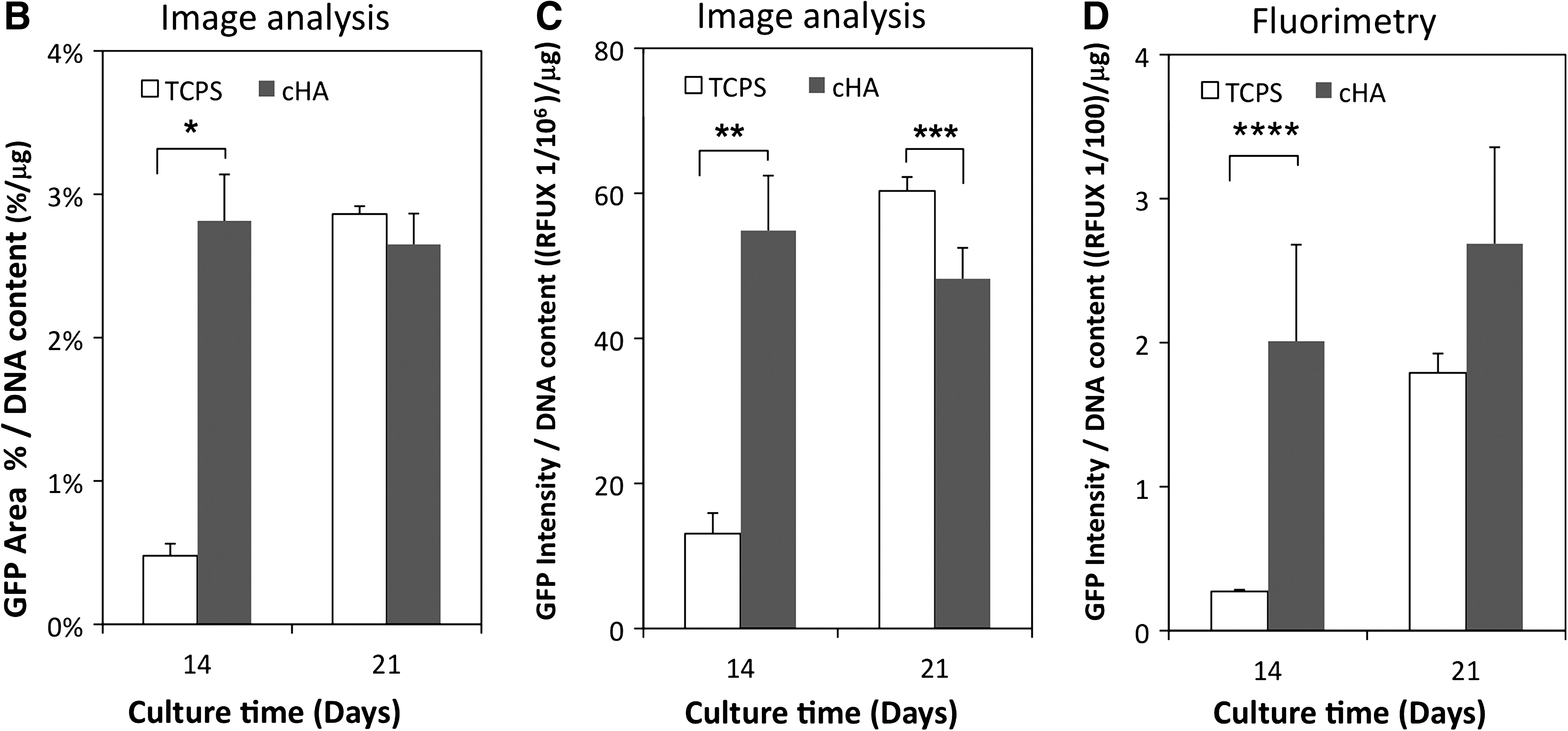
The quantitative measure of the percent of culture area covered by GFP-positive cells at 14 and 21 days was determined from fluorescence microscopy. At 14 days, there was approximately fourfold more GFP-positive area on the cHA than on TCPS (15.8 ± 1.8% vs. 4.1 ± 0.7%, p = 0.0005). After normalization to total cell number as measured by μg of DNA/well (Fig. 2B), the cHA values became relatively larger (p = 0.0002) due to fewer cells present on the cHA than TCPS (cHA had 65% and 78% of the cell number on TCPS at 14 and 21 days, respectively). At 21 days, before normalization to total cell number, the TCPS had a larger GFP-positive area than cHA (23.8 ± 0.5% vs. 17.2 ± 1.4%, p = 0.009), but after normalization to cell number the difference was not statistically significant (p = 0.285). The results from the area analysis and the intensity analysis were generally consistent: normalized mean intensity of GFP on cHA determined by image analysis was fourfold higher and statistically greater than TCPS (p = 0.0009) at 14 days (Fig. 2C). At 21 days, normalized TCPS mean intensity as determined by image analysis was 1.25 higher (p = 0.035) than cHA.
Fluorimetry data were consistent with image analysis results and showed that GFP intensity increased with time on both surfaces, reflecting increased differentiation level toward the osteoblastic phenotype (Fig. 2D). cHA accelerated the differentiation of the cells to osteoblasts as evidenced by enhanced GFP expression at 14 days relative to TCPS. Normalized mean intensity values determined by fluorimetry were approximately sevenfold more for cHA than TCPS (p = 0.0002). The correspondence of fluorimetry analysis, which analyzed the whole well, with image analysis, which used 36 sampling points, further proves that the image analysis area was representative of the entire well.
Imaging and fluorometric intensity of XO-stained mineralized deposits
Mineralized matrix deposition by the osteoblasts was observed after XO staining by microscopy. At day 14, the mineral deposits on TCPS surfaces were in discrete high cell density nodules (Fig. 3C). While GFP expression on cHA was present over a larger area and more intense (Fig. 3F), the mineral deposition (Fig. 3G) was barely visible in the images taken at day 14 using the exposure times established from 21-day TCPS cultures. Importantly, what was visible was associated with GFP-positive cells, indicating that the mineral is osteoblast initiated. The XO absorption by the cHA made it difficult to distinguish cell-deposited mineral from the mineral (cHA) substrate using image analysis, particularly in the concatenated images taken at 10× magnification. However, at 21 days and at higher magnification (Fig. 4B), the XO-stained cell-deposited mineral could be distinguished visually because it had a brighter, lacy-appearance that was distinct from the low level, pebbly appearance of the cHA (Fig. 4A). Importantly, this XO-stained lacy morphology was found only in association with differentiated GFP-positive osteoblasts and not scattered throughout the plate, which would have indicated non-cell-mediated precipitation.

Tri-color fluorescent images of the mCOBs shown as separate channels and merged, of 14- and 21-day mCOB cultures stained with DAPI and xylenol orange (XO) (

Fluorescent microscopy images after xylenol orange staining of (
Since the XO-stained cHA features could not be completely thresholded out of the images due to overlap with the deposited mineral, the amount of XO staining was quantified by fluorimetry only because the biomaterial background obtained by fluorimeter readings could be simply subtracted. The mean fluorescence intensity quantified by fluorimetry was found to be ninefold higher on cHA than on TCPS (p < 0.000004) at 21 days.
Discussion
In this study we have demonstrated that osteoprogenitor cells from transgenic mice harboring the collagen I Col1a1-promoter-driven transgene pOBCol2.3GFP5,6 can be used to distinguish in vitro differentiation and mineralization patterns on CaP-coated disks and TCPS. This is the first usage of osteoblast-reporter cells for nondestructive, quantitative screening the osteopromotive capabilities of biomaterials. Col2.3GFP expression is quite specific to a late-stage mineral-producing osteoblast,5–7 and therefore these cells are a useful tool for analyzing the effect of biomaterials on osteoblast differentiation. The main advantage of using these cells is that it is possible to forgo destructive measurements of fluctuating biochemical markers, and conduct a simple visual, nondestructive fluorescent assay to assess osteoblast differentiation status during cell culture on biomaterial substrates. Because the pOBCol2.3GFP cells do not have to be removed from the biomaterial to assess their differentiation status, important spatial information, for example, correlation of osteoblast location to biomaterial structural features, and effects of cell–cell interactions are retained with this method.
In this study, interestingly and most importantly, the relatively homogeneous nature of the differentiation process on cHA was clearly observed for the first time by fluorescent microscopy of live cultures and was in great contrast to the typical heterogeneous or nodule associated differentiation on standard TCPS. 31 The continuous in vitro mineralization pattern observed herein mirrors the direct bone bonding and lack of fibrous tissue that is seen during successful in vivo surgical implantations of HA- and cHA-coated orthopedic devices.32,33 This in vitro–in vivo correlation indicates that the technique has value as a preclinical screening device for orthopaedic applications of biomaterials. If fluorimetry measurements alone had been used, the unique response of the cells to the cHA would not have been evident. Fewer data points that represent averages of larger areas are obtained with the fluorimeter due to nature of the technique; thus, image analysis is recommended when assessing GFP-lineage reporter cell expression in in vitro assays. However, due to the greater dynamic range of the photomultiplier in the fluorimeter than in the fluorescent microscope's camera, the differences between XO-stained mineral and XO-stained cHA substrate were much easier to determine with the fluorimeter. Automated segmentation of cHA from mineralized matrix by visual thresholding was not possible using image analysis because the cHA background overlapped with the XO-stained mineral.
Our study indicates that the cHA provides a cue for a direct and efficient conversion of monolayer cultures to osteoblasts without the need for the creation of a high-density three-dimensional nodule of cells that is the classic in vitro mineralization mode on tissue culture plastic. The exact mechanism of how cHA enhances bone formation remains to be elucidated, but has been shown or postulated to be an effect of (1) selective cell attachment in which only committed cells attach to the cHA scaffold surface, 34 (2) concentrated absorption and preferred presentation of cell adhesive proteins that engage and activate integrins associated with differentiation,18,35,36 (3) the crystalline topography, which reduces focal adhesions, cell spreading, motility,15,34 and proliferation rate, 21 and (4) increased free phosphate levels. 17 Recently, it has been shown that mesenchymal stem cells cultured on cHA substrates led the cells to generate potent inductive substances released into the medium that induced mineralization when the conditioned medium was used to culture other cells. 25 Our previous cell attachment studies on cHA and TCPS showed that both primary cells, and cells from an established cell line, attach in lower numbers to the cHA than TCPS: 23 ± 2% on cHA vs. 35 ± 2% on TCPS for the primary cells, and 49 ± 3% on cHA vs. 83 ± 8% on TCPS for the cell line. 27 Yet, only rarely was a dead (red) cell observed on either surface with a live/dead stain, indicating that decreased attachment was not due to toxicity of the cHA, but rather a selective attachment mechanism. A recent atomic force microscopy study indicates that while less fibronectin adsorbs to cHA, the cell binding domain of fibronectin is more available on cHA than other surfaces, such as gold. 37 This further supports a selective cell attachment on cHA in which more cells with α5β1 integrin receptors attach to the surface leading to enhanced differentiation and slowed proliferation, similar to that reported for cells on collagen coatings.38,39
Typical alkaline phosphatase and von Kossa–stained cultures are destructive endpoint assays that lack the specific information made possible by use of the osteoblast lineage reporter transgenic mouse cells combined with nondestructive XO staining. Nonphysiological mineral deposition that can occur during in vitro osteogenesis assays of multipassaged mesenchymal stem cells 40 can be easily distinguished from cell-mediated mineral in these cultures by confirming the presence of both the Col2.3GFP and the XO in mineralized deposits. von Kossa cannot be used to assess mineralization when the cells are grown on CaP-containing biomaterials due to high levels of nonspecific absorption to the CaP. We have demonstrated here that XO can be used to visually distinguish cell-produced mineral from synthetic CaP and that the differences can be quantified using a fluorimeter and subtraction of background from nonmineralizing cultures on the substrates. The fact that XO is not toxic to cells at the concentrations needed to stain the mineral is an additional advantage because the same cultures can be assessed at multiple time points without termination.
Most techniques are incapable of focusing on a defined cellular population within a heterogeneous culture environment and often treat cells as uniform population for easier assessment. The use of tissue-specific reporters overcomes this limitation by better defining cell types without requiring sample destruction. It is a technology of high utility, particularly for those that are unfamiliar or untrained in the biochemical assays routinely utilized by molecular and cell biologists to identify a mature osteoblast. At the same time, interpreting reporter gene expression is not left without its challenges. For example, the fluorescence from pOBCol2.3GFP cells comes on quickly as compared to the pOBCol3.6GFP cells 5 and is brightest during initial mineral deposition, yet shows decreased reporter expression thereafter. This is the reality of in vitro osteogenesis, in which mature bone cells possess varying levels of metabolic activity, and eventually become apoptotic before or after being embedded within a dense mineralized matrix. 41 Observations of Col2.3GFP expression at multiple time points in conjunction with observation of XO-stained mineralized matrix are therefore recommended to correctly interpret the extent of in vitro osteogenesis since the mineralized matrix will remain throughout the culture time period. Because cells differentiate in an asynchronous manner within the plate, large areas of the plate should be analyzed to obtain accurate values.
Exposure settings are a critical parameter in studies such as these because overexposure leads to loss of high gray-scale values and prevents accurate image analysis. It is important to set camera exposure times low enough to capture the highest expression levels in mature 21-day cultures and then use those settings consistently throughout the studies. This also avoids over-enhancement of the image and the premature identification of Col2.3GFP-positive late-stage osteoblasts. The use of fluorescent beads as a constant, unchanging standard measured at every time point would be an enhancement to the analysis technique because it would account for variation in microscope source intensity (by normalization of data obtained from the plates to the beads). In these experiments the two different culture substrates were read on the same day, so the comparisons reported are valid.
For a more in-depth analysis, it is possible to remove the cells from the biomaterial scaffold and sort the GFP-positive cells by flow cytometry for a highly specific study of receptor or gene expression changes in response to being cultured on various biomaterials. We found, however, that it was difficult to fully remove all of the osteoblasts from the cHA and the TCPS once they had deposited a mineralized matrix. This was reflected by our previously reported large standard deviations of total DNA at day 14 and 21 time points. 27 The cells cultured on cHA deposited a mineralized matrix sooner than cells on TCPS and thus had larger standard deviations for total DNA than TCPS at earlier time points (e.g., 14 days: 5.59 ± 2.02 μg/well for cHA vs. 8.47 ± 0.04 μg/well for TCPS). By 21 days the standard deviations were similar.
These novel lineage-reporter cells should not be confused with previously reported GFP-labeled cells from transgenic mice that are initially GFP-positive and remain GFP-positive throughout a study and are not associated with the cell's lineage status.2,42 The Col2.3-associated GFP used in the present study is only expressed by mature osteoblasts capable of secreting a mineralized matrix and osteocytes, and not early osteoprogenitor cells or other cell types. Other fluorescent lineage reporters have been developed for earlier and later stages in the osteoblast lineage8,43 and could be used for screening effects of other factors intended to affect immature progenitor cells. Cellular response to growth factor/biomolecule stimulation can be more easily and effectively studied with these fluorescent lineage reporter cells. 7 While we utilized cells obtained from calvaria of the pOBCol2.3GFP transgenic mice, mesenchymal stem cells obtained from the marrow or fat of pOBCol2.3GFP transgenic mice can also be utilized in a similar manner. 6 GFP reporters for other musculoskeletal cell types such as chondrocytes and muscle have recently been developed as well, which can allow precise mapping of lineage progression of multiple cell types on or within scaffolds.44,45
Conclusions
We report on a new in vitro method to assess the effect of biomaterials on the differentiation of osteoprogenitor cells. The new method utilizes osteoprogenitor cells from transgenic mice harboring the Col1a1-promoter-driven transgene pOBCol2.3GFP that only expresses GFP upon differentiation to a late-stage bone cell. Importantly, this technique is superior to traditional alkaline phosphatase/von Kossa staining because it allows for real-time assessment of cell lineage status and patterns of mature osteoblasts, and also allows cell-initiated mineral deposition to be discriminated visually without terminating the culture. This technique clearly revealed the nonhomogeneous nature of osteoblast differentiation on TCPS and the more uniform differentiation on cHA. The cell signaling mechanism provided by the cHA substrate did not require that the cells form the discrete nodule or multicell aggregates typically observed during osteoblast differentiation on TCPS. Because this technique can assess area percentage of in vitro bone formation, it will be highly useful for preclinical screening of biomaterials intended to promote enhanced bone area contact associated with improved oseointegration. The GFP lineage reporter method is rapid and efficient and provides new information about the differentiation behavior of cells on biomaterials because spatial information is retained.
Footnotes
Acknowledgments
This material is based upon work supported by the State of Connecticut under the Connecticut Stem Cell Research Grants Program and the U.S. Army Medical Research and Material Command. The authors would like to acknowledge and thank Dr. David Rowe of the University of Connecticut Health Center for supplying the pOBCol2.3GFP lineage reporter mice.
Disclosure Statement
No competing financial interests exist.