
Abstract
After surgery, wound healing begins with a well-orchestrated integration of several cytokines, cells, and extracellular matrix. Some studies show an involvement of stem cells in wound healing. However, little is known about the mechanism that leads to the migration of stem cells. Wound fluid (WF) with its cytokines may play an important role. We investigated in the present study the in vitro effects of WF on adipose-derived stem cells (ADSCs). Survival, proliferation, structural integrity, changes in the multidifferentiation potential, and surface markers (cluster of differentiation [CD] 105, CD73, CD90) of ADSCs after cultivation with WF was analyzed. Further, the migration effect of WF on ADSCs was evaluated. The proliferation rate and the migration potential of ADSCs were enhanced significantly by cultivation with WF. There was also a change in the quantity of surface markers after cultivation with WF. In conclusion, in vitro expansion of stem cells with WF proved possible. WF and its cytokines could represent one primary reason for the migration of stem cells toward the wound. Future investigation is warranted to clarify the significance of the shift in surface markers.
Introduction
Inflammation, proliferation, and remodeling constitute wound healing. 2 In addition, cytokines take part in cellular growth and cellular repair as well as protein synthesis. 3 Some in vitro and in vivo studies show an involvement of multipotent stem cells in wound healing.4,5 Mesenchymal stem cells (MSCs) from bone marrow and adipose tissue are well-established cell sources for tissue regeneration. The characterizations of stem cells are undifferentiation, the ability of self-renewal, and high proliferative capacity. 6
In the clinical practice, bone marrow-derived stem cells (BMSCs) may be less satisfying. For example, an invasive procedure is needed to get enough BMSCs from a donor. The cell number and differentiation potential differ between young and mature donors as well.7–9 Simple surgical procedures to obtain adipose tissue, for example, by liposuction and the uncomplicated enzyme-based isolation procedure, make adipose-derived stem cells (ADSCs) attractive for clinical use. 10 ADSCs have the multipotential capability to differentiate under specific conditions into different lineages of mesenchymal tissues such as bone, muscle, fat, and cartilage. 11 Stem cells from bone marrow, adipose tissue, and umbilical cord blood are not different in terms of their differentiation capacity.12,13 There are no specific cellular markers for identifying adult stem cells, but the availability of cell surface markers combined with their differentiation potential into different mesenchymal tissues in vitro are established analyses for identifying stem cells. 14 Such markers include cluster of differentiation (CD) 105, CD73, and CD90. BMSCs do not express CD34, CD45, CD14 or CD11b, CD79a or CD19, and HLA class II. 15 In contrast to BMSCs, ADSCs are CD34 positive but CD31 negative. 16 BMSCs have some immunosuppressive properties in vitro and in vivo. 17 Finally, ADSCs have shown certain immunosuppressive properties in vitro as well. 18 How stem cells migrate to the site of injury has yet to be elucidated. Tissue-specific homing of leukocytes may depend on cytokines, tissue-specific adhesion molecules, and homing receptors. 19 Several studies have reported on the expression of chemokine receptors and chemokines in BMSCs.20,21 However, little is known about the mechanism of stem cell migration and the interactions of chemokine receptors in regeneration of damaged tissue.
The objective of the present study was to analyze the in vitro effects of wound fluid (WF) containing cytokines on ADSCs. Thus, the study focused on survival, structural integrity, and differentiation of ADSCs, as well as on migration effects of WF on ADSCs. Moreover, the effects of various WF concentrations on the proliferation of ADSCs were analyzed and compared with fetal calf serum (FCS). This work aims at contributing to a deeper understanding of the process of wound healing, the importance of WF and its cytokines, and the intrinsic role of stem cells.
Materials and Methods
In the present study, different analytic measures were applied to focus on the effects of WF on ADSCs in vitro.
Wound fluid
Collection and purification
WF was collected from a vacuum drain of patients who underwent a planned neck dissection at the Department of Oto-Rhino-Laryngology, Plastic, Aesthetic, and Reconstructive Head and Neck Surgery, University of Wuerzburg, Germany. In accordance with creating identical experimental issues, only WF from the neck was included in the present study. WF was centrifuged immediately at 1300 rpm for 10 min at 4° C to reduce cell debris. To remove immune cells, a second centrifugation in Leucosep medium (Ficoll Paque Plus GE Healthcare, Freiburg, Germany) was conducted followed by another filtration using a 0.45 μm syringe filter (Sarstedt, Nümbrecht, Germany). To avoid bacterial infection, 100 units/mL of penicillin and 100μg/mL of streptomycin (1% penicillin/streptomycin) was added. Finally, WF was stored at −80°C until use.
Cytokine assay
To detect the presence of different cytokines in WF, Dulbecco's modified Eagle's medium (DMEM), and FCS, the dot blot assay representing a semiquantitative method (RayBiotec, Inc., Norcross, GA) was performed. For the detection of cytokines, we used the manufacturer's protocol. First, WF was added to the membrane. The membrane was spotted with antibodies against different cytokines. Next, blocking buffer was added to the membrane for 30 min. Thereafter, several washing steps followed. Another 2 h of incubation with biotin-conjugated antibodies and horseradish peroxidase-conjugated streptavidin was investigated. After adding the detection buffer and exposure to an X-ray film, the proteins were observed. The cytokines are represented as dots with different intensity and growth size.
Isolation and culture of ADSCs
Human ADSCs were isolated from subcutaneous adipose tissue of healthy donors undergoing liposuction for abdominoplasty or gynecomastia surgery. Studies were approved by the Ethics Committee of the Medical Faculty, University of Wuerzburg (12/06), and informed consent was obtained from all individuals included in the study. The isolation procedure was performed according to the methods described in previous studies. 16
Briefly, adipose tissue was extensively washed with phosphate-buffered saline (PBS) (Roche Diagnostics GmbH, Mannheim, Germany) and enzymatically digested at 37°C for 3 h with a concentration of 15 mg collagenase P (Roche Diagnostics GmbH) per 100 mL lipoaspirate. To obtain the cell pellet, several washing and centrifugation steps were performed. The pellet was resuspended and passed through a 100μm cell strainer (BD Bioscience, Heidelberg, Germany) to remove debris. Cells were plated in culture flasks at 37°C/5% CO2. Expansion medium (EM) consisted of DMEM (Gibco Invitrogen, Karlsruhe, Germany), 10% FCS (Linaris, Wertheim–Bettingen, Germany), and 1% penicillin/streptomycin. After 24 h, ADSCs were rinsed with PBS to remove residual nonadherent cells. Medium was changed every other day. When cells reached >70% confluence (P0), cells were trypsinized with 0.25% trypsin (Sigma-Aldrich, Schnelldorf, Germany), resuspended in EM, and subcultured at a concentration of 2000 cells/cm2. Cell morphology was then analyzed by microscopy (Leica DMI 4000B Inverted Microscope; Leica Microsystems, Wetzlar, Germany).
In vitro differentiation
Differentiation procedures were performed to analyze the multidifferentiation potential. ADSCs were cultivated with WF and EM for 1 week. The differentiation into 3 mesenchymal tissues was done according to the following protocols.
Osteogenic differentiation
Osteogenic differentiation was performed in monolayer culture with 1×104 cells until 70% confluence was reached. The osteogenic induction medium contained EM, 10−7 M dexamethasone, 10−3 M β-glycerophosophate, and 2−4 M ascorbate-2-phosphate (all Sigma-Aldrich) as described by Pittenger et al. 6 Every third day media was changed. As a negative control, cells were maintained in EM. The von Kossa staining detected an osteogenic differentiation stage. It was performed according to the protocol previously described. 22 Cells were washed with PBS without Ca++ and Mg++ thrice and were fixed with 70% ethanol for 20 min at room temperature (RT). After washing, the cells were stained according to the protocol for 60 min with 5% silver nitrate (Sigma-Aldrich) under ultraviolet light (254/366 nm; Desaga Lightbox UVIS, Burladingen, Germany) at RT. Next, cells were fixed with 5% sodium thiosulfate (Applichem, Darmstadt, Germany) for 5 min. Finally, cell nuclei were stained with Nuclear Fast Red. Von Kossa staining is routinely used to characterize mineralization by staining the calcium mineral component dark brown.
Adipogenic differentiation
Adipogenic differentiation was performed in monolayer culture as described by Pittenger et al. 6 Cells were plated at a density of 1–2×104 cells/cm2. Adipogenic differentiation was induced by EM, 10−7 M dexamethasone, and 10 ng/mL recombinant human insulin. Adipogenic cell differentiation was confirmed by staining with Oil Red O to show the presence of intracellular lipid droplets.
Chondrogenic differentiation
For chondrogenic differentiation, the pellet culture system was used. The cell pellets were cultured in defined chondrogenic differentiation media (Lonza, Basel, Switzerland) and by adding 10 ng/mL transforming growth factor-beta 3. The medium was replaced every other day for 3 weeks. After 21 days, the pellets were embedded in Tissue-Tek® O.C.T.™ Compound (Optimal Cutting Temperature Paraffin, Tissue-Tek®). The cryosections were stained with Alcian blue to show the presence of glycosaminglycane.
Immunohistochemistry
ADSCs were fixed with 4% paraformaldehyde for 30 min at RT. Another 5 min of fixation with acetone at RT was followed. Cells were blocked with 10% bovine serum albumin (BSA) (Carl Roth GmbH, Karlsruhe, Germany) in Tris-buffered saline 200 mM Tris-base (pH 8), 8% NaCl, and 1% Tween-20 (TBS-T) (Sigma-Aldrich). Cells were maintained in TBS-T containing 1% BSA and with a mouse monoclonal antibody against α-tubulin (1:500; Sigma-Aldrich) for 10 h. After three washing steps with TBS-T (each step 5 min), cells were incubated for 1 h in 1% BSA at RT with Alexa 488 goat anti-mouse secondary antibody 1:500 (Gibco Invitrogen) and 5μg/mL DAPI (Sigma-Aldrich).
Effects of WF on ADSCs
After the incubation of ADSCs with WF for 1 week, we examined the surface markers using flow cytometry, as well as changes in the proliferation and multidifferentiation capacity, the migration effect, and the ability for in vitro wound healing.
Flow cytometry
ADSCs were incubated with WF in a 12-well plate for 1 week and were analyzed by flow cytometry (BD FACSCanto™; BD Bioscience). Cells were incubated with anti CD105, anti CD73, anti CD90, and anti CD31. Flow cytometric analysis was performed with and without antibody staining. As a control, cells were incubated with EM.
Proliferation analysis
About 1×104 ADSCs were incubated with WF, EM, and DMEMW/O at 37°C with 5% CO2 for 5 days, while electronically counting the cell number each day (Casy® Technologies, Innovatis AG, Reutlingen, Germany). Further, the proliferation of ADSCs was investigated depending on different WF concentrations (1%, 10%, 15%, 30%, and 50%). The proliferation was analyzed by staining with crystal violet, and the absorbance was measured with Titertek Multiskan PLUS (MK II) ELISA-reader (Labsystems, Helsinki, Finland) at 570 nm.
Migration assay
Transwells with 6 μm membrane pores (Nunc, Langenselbold, Germany) were coated with 50% extracellular matrix gel (Sigma-Aldrich) for 30 min. About 1×105 ADSCs were coated on the top surface of the membrane and incubated with DMEM for 24 h at 37°C with 5% CO2. Subsequently, WF was added to the bottom of the chamber, followed by incubation for 24 h. To remove nonmigratory cells on the upper membrane, a cotton swab was used. Migrated cells that were on the lower surface of the membrane were stained for 25 min with 1% crystal violet (Sigma-Aldrich) in 0.1 M borate and 2% ethanol. After one washing step with aqua destillata, cells were incubated with 10% acetic acid for 20 min. The measurement was performed using the multi-plate reader (Titertek Multiskan PLUS (MK II); Labsystems) at a wavelength of 570 nm.
Wound healing
The scratch assay was performed in monolayer culture in a six-well plate (Nunc). Cells were incubated for 24 h at 37°C at a concentration of 1×105 cells/mL and culture medium containing WF with 1% penicillin/streptomycin. Control cells were incubated with EM. A straight line wound was induced with a sterile 1 mL pipette tip. By washing with PBS, cellular debris was removed. Culture plates were incubated for 24 h at 37°C with 5% CO2. To analyze the relative migration, plates were photographed (Leica Microsystems), and the number of ADSCs that migrated into the wound area was counted and compared between WF and EM.
Statistics
For the statistical analysis, GraphPad Prism software (version 4.0) was used. First, we analyzed whether the distribution was Gaussian. In the case of Gaussian distribution, unpaired t-test was used; otherwise, Kruskal–Wallis test was performed. Statistical significance (p<0.05) is indicated in the figures.
Results
Cytokine assay
To evaluate the availability of different cytokines, a semiquantitative cytokine assay was performed. WF contained a variety of cytokines, for example, responsible for inflammation (tumor necrosis factor-α, -β, interleukin [IL]-6, and Oncostatin-M), anti-inflammation (IL-10), chemotaxis (monocyte chemotactic protein [MCP]-1, MCP-2, MCP-3, and IL-8), angiogenesis (Angiogenin), and growth factors (vascular endothelial growth factor, insulin-like growth factor-1, IL-7, growth-regulated oncogene [GRO], and GRO-α platelet-derived growth factor-BB) (Fig. 1). DMEM as negative control contained epidermal growth factor. The positive dots in the DMEM group demonstrated that the assay was correctly performed (Fig. 1C).

Representative dot-blots of
ADSC isolation
ADSCs were isolated according to a standard technique for the isolation of cells from adipose tissue. Microscopic analysis revealed that cells treated with EM demonstrated the typical spindle-shaped, fibroblast-like morphology. There was a morphological difference between cells cultivated in WF compared with cells cultivated with EM (Fig. 2): ADSCs cultivated with WF were smaller and had a denser cytoplasm compared with ADSCs cultivated with EM. There was no structural instability or negative influence on viability during the cultivation of cells with WF or EM.

Microscopic illustration of ADSCs cultured with WF, DMEM, and FSC.
ADSC differentiation
The potential of ADSCs to differentiate into osteocytes, adipocytes, and chondrocytes after cultivation in WF was confirmed by von Kossa, Alcian blue, and Oil Red O staining (Fig. 3).

ADSCs were cultured in WF for 1 week. After 1 week, the potential to differentiate into osteocytes, adipocytes, and chondrocytes was confirmed by the von Kossa, Alcian blue, and Oil Red O staining.
Flow cytometry
Cells were tested with flow cytometry for the presence of ADSC typical surface markers CD105, CD90, and CD73. CD31 was a negative control. During the cultivation with WF, there was a change in the quantity of CD105, CD90, and CD73. Cells with both surface markers CD90 and CD73 were attenuated during cultivation with WF, whereas the expression of CD90 alone was enhanced. The expression of CD73 alone was nearly 0% during cultivation with WF (Fig. 4).
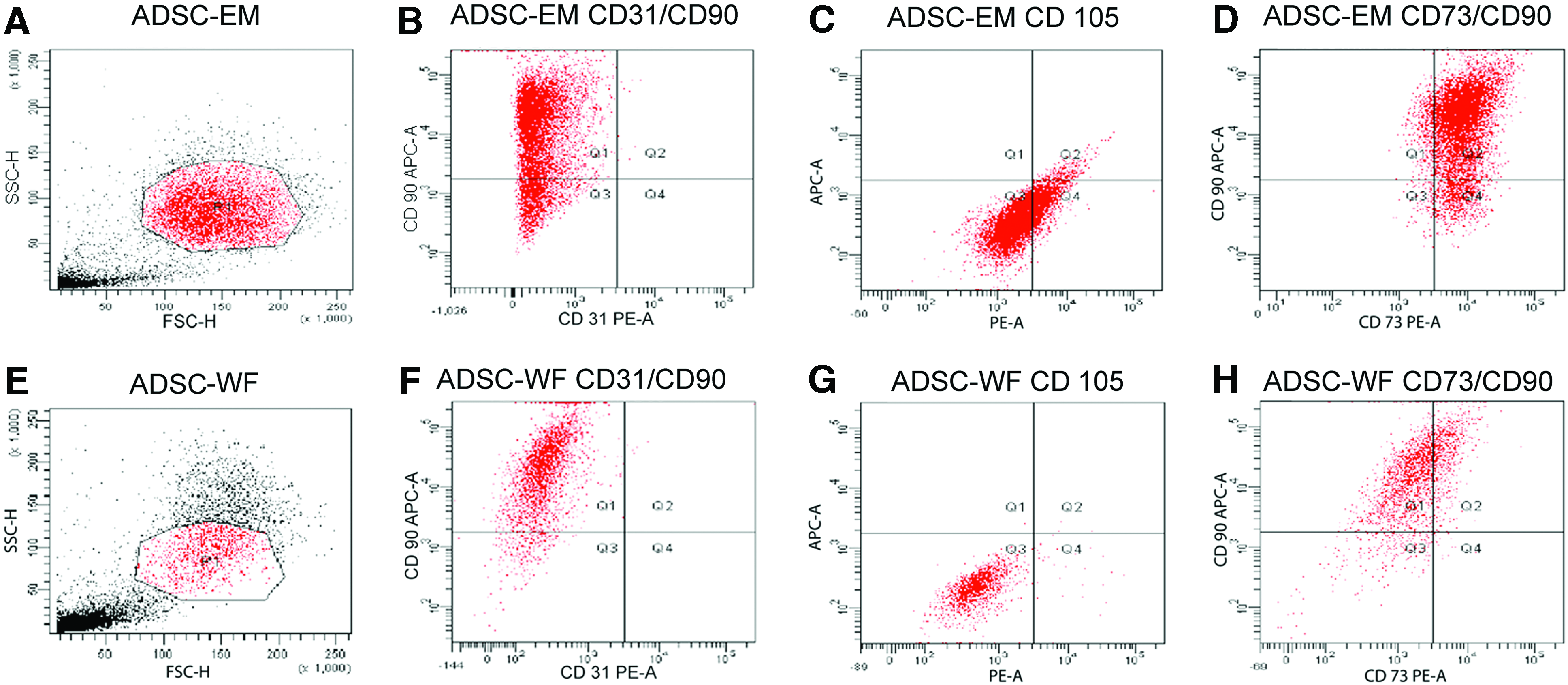
Flow cytometric analysis of surface markers routinely used for characterization of human ADSCs.
Cell proliferation assay
Cell proliferation was increased by cultivation with WF. The difference was statistically significant, with a p-value <0.001 analyzing curves at all time points (Fig. 5). To investigate the existence of a concentration-dependent relationship for cell proliferation, ADSCs was cultivated with increasing WF concentrations (1%, 10%, 15%, 30%, and 50%). Results indicated that the proliferation rate was indeed dependent on WF concentration, with the highest cell proliferation rate observed at a concentration of 50% WF (Fig. 6).
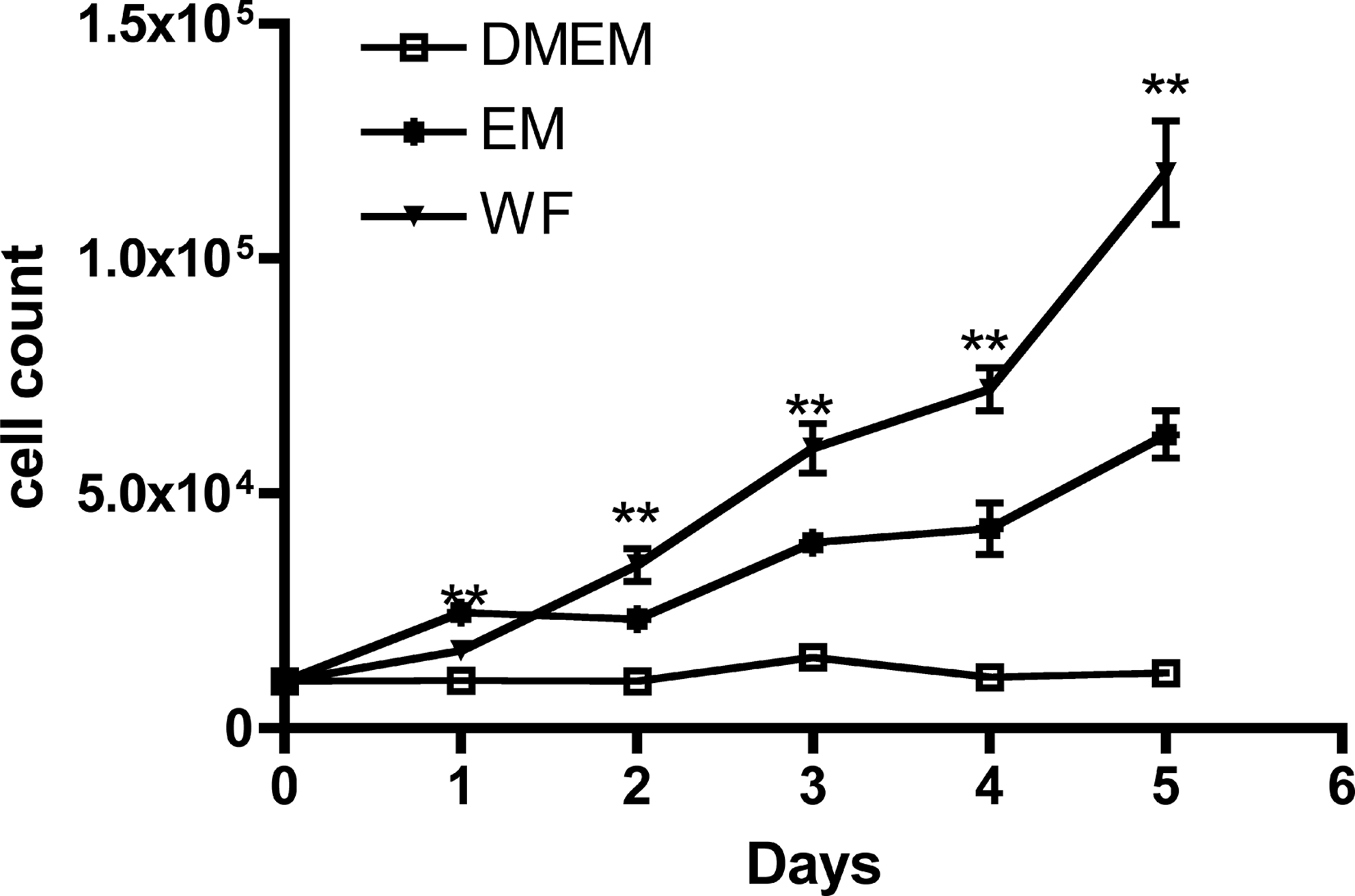
The effect of WF on cell proliferation was analyzed by cell counting. About 104 cells per well were incubated with WF, EM, and DMEM in a 24-well plate, and each day the cell count was measured. Statistical differences are indicated as **p<0.001.

The concentration-dependent increase in cell proliferation was examined by cell staining with crystal violet and the absorbance measurement at 570 nm. About 104 cells/well were cultured in a 24-well plate with different WF concentrations. Statistical differences are indicated as *p<0.05; **p<0.001.
Transwell migration of ADSCs
The transwell migration assay was investigated to study the migration effect of different media (WF, EM, and DMEMW/O) on ADSCs. The migration of ADSCs cultivated with WF was significantly (p<0.001) higher than in EM and in DMEMW/O (p<0.001). There was also a significant difference (p<0.001) in cell migration between DMEM containing 10% FCS and DMEM without supplements (Fig. 7).
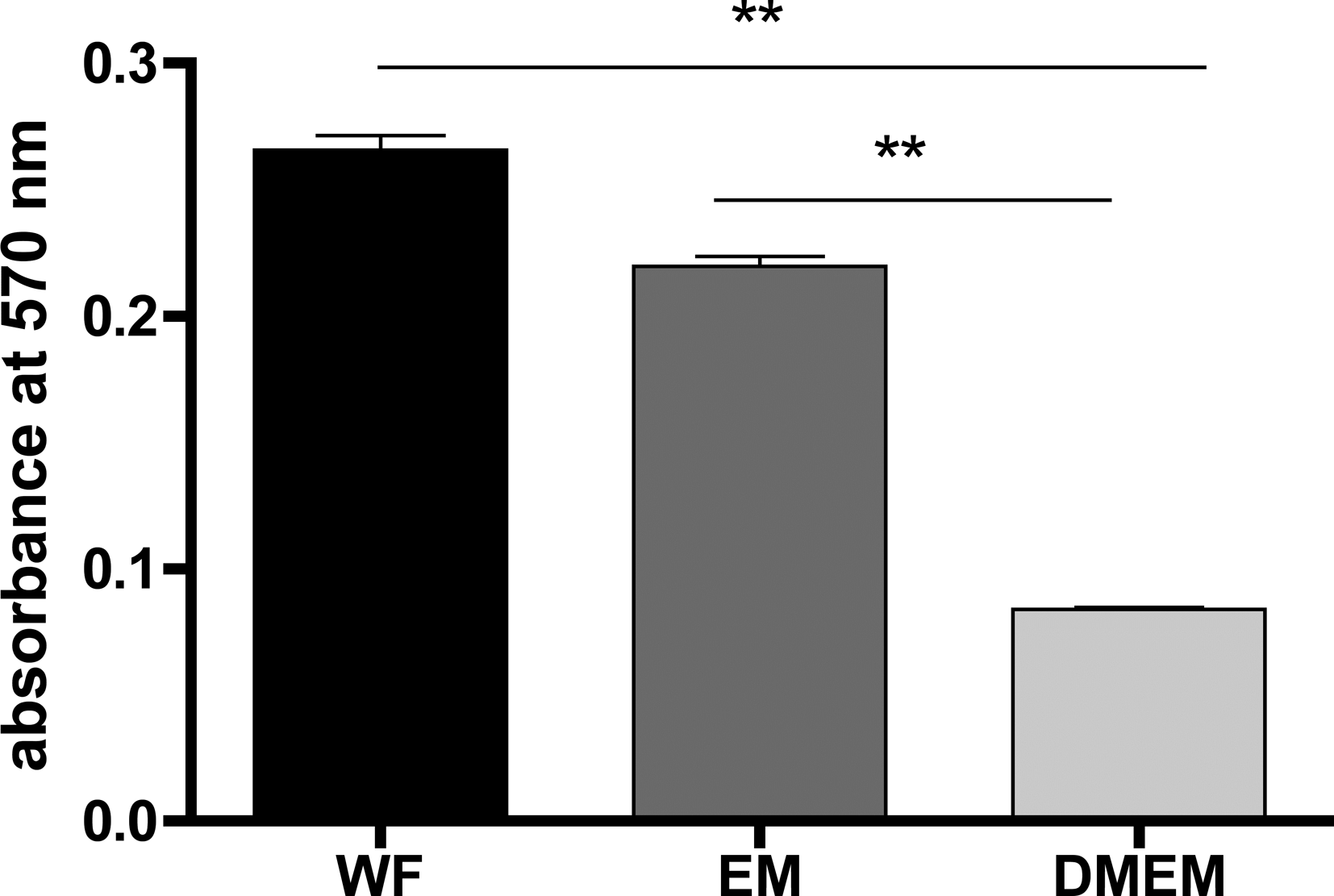
ADSCs were coated on the top surface of the membrane and incubated with DMEM for 24 h at 37°C with 5% CO2. WF, EM, and DMEMW/O were added to the bottom of the chamber followed by incubation for 24 h. Cell migration was analyzed at 570 nm by the Titertek Multiskan PLUS (MK II) ELISA-reader. Statistical differences are indicated as **p<0.001.
Wound healing
The scratch test is commonly used to test migration of cells toward a wound that has been created in a cell monolayer. After a repair period of 24 h at 37°C and 5% CO2, ADSCs cultivated with both media (WF and EM) migrated into the wound area. The migration of ADSCs cultivated with EM into the wound appeared slightly higher than the number of ADSCs cultivated in WF. However, this difference proved not to be statistically significant (Fig. 8).
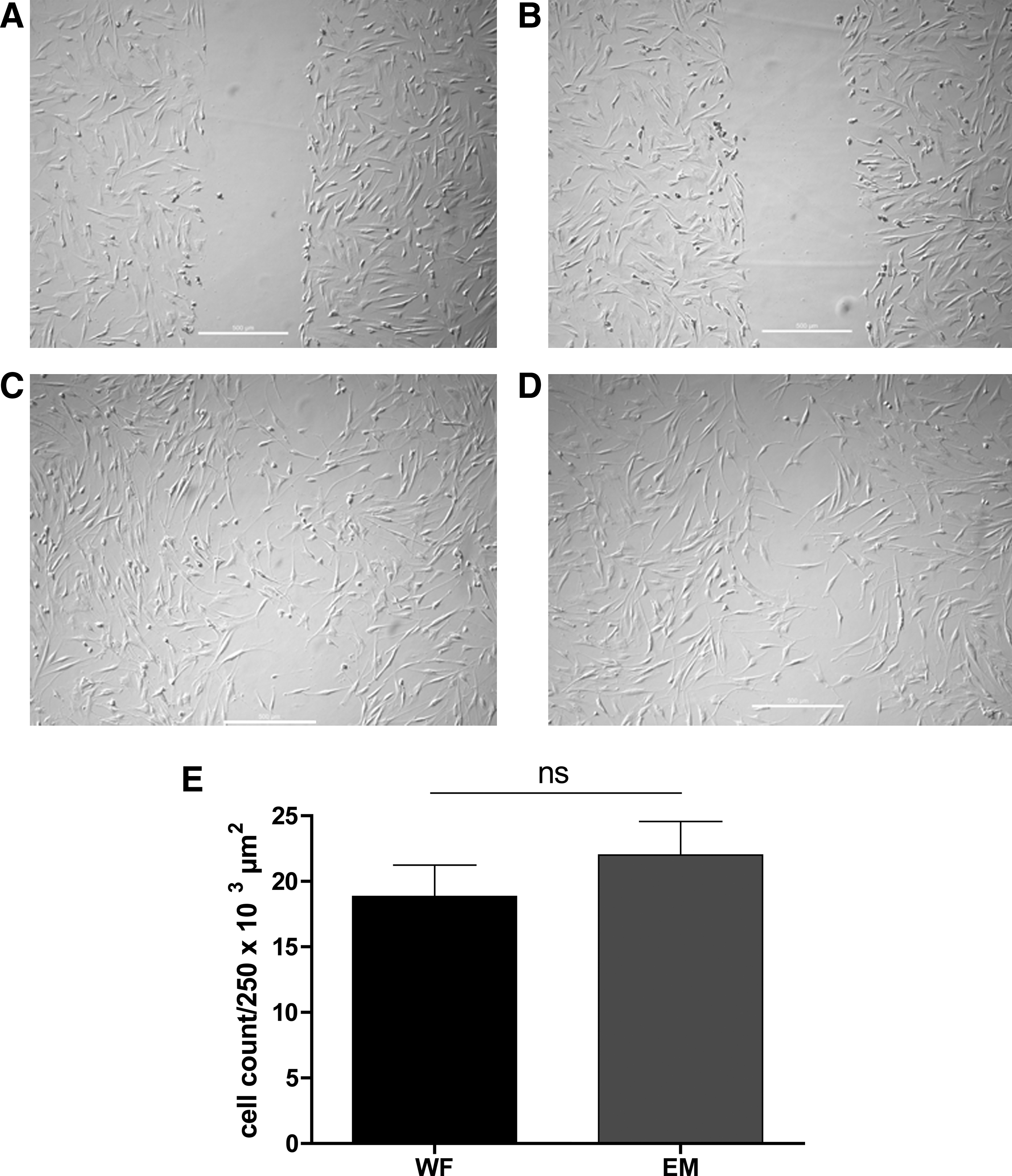
ADSCs were cultured in EM and WF to a nearly confluent monolayer. A wound was generated by a sterile plastic pipette tip (t=0 h). To analyze the relative migration, plates were photographed after 24 h, and the number of ADSCs that migrated into the wound was counted and compared between WF and EM.
Discussion
The objective of this study was to analyze the in vitro effects of WF on ADSCs to better understand the mechanism of stem cell involvement in wound healing. Inflammation, proliferation, and remodeling constitute wound healing. 23 Some in vitro and in vivo studies have shown the participation of multipotent stem cells in wound healing.4,5 Further, cytokines modulate immune and repair processes by controlling cellular growth, differentiation, metabolism, and protein synthesis. 3 In our study, ADSCs showed a significant enhancement of cell proliferation in the presence of WF. Kakudo et al. investigated the effects of platelet-rich plasma on the proliferation of ADSCs and found that 5% of platelet-rich plasma promoted maximal cell proliferation, whereas higher concentrations did not do so. 24 In contrast to these findings, our results indicated that the proliferation of ADSCs was enhanced significantly with increasing concentration levels of WF, thus indicating a dose-dependent relationship.
In particular, the two growth factors PDFG and transforming growth factor-β are important for normal tissue repair processes.25,26 MSCs, found in many adult tissues, for example, bone marrow and adipose tissue, are frequently used in tissue regeneration, for example, in skin wound repair, bone repair, cerebral ischemia, or in heart diseases.27–30 However, the mechanism of homing stem cells to the side of injured tissue is not yet fully understood.
The migration of stem cells to the site of injury depends on several aspects. Cytokines and cytokine receptors might play an important role in these processes. Chemokine (C-C motif) ligand 5, also called CCL5/RANTES, seems to be the most potent chemokine for ADSC migration. 31 Further, the CXCR4 receptor (CXC chemokine receptor) also plays an important role in the migration and homing of ADSCs. 32 According to Kroeze and colleagues, ADSCs express chemokine (C-C motif) receptor 1 (CCR1), CCR 3, CCR 4, CCR 6, CCR 7, CCR 10, and CXR 1–4.23,31 Indeed, RANTES belongs to the rapidly growing chemokine family and induces leukocyte migration. 33 WF and its cytokines, including CCL5/RANTES, are potent chemoattractants. The migration of ADSCs toward WF in transwell chambers was significantly enhanced compared with EM and DMEMW/O. The migration of EM was also significantly improved compared with DMEMW/O. DMEMW/O does not contain any cytokines or growth factor. This was shown semiquantitatively in our dot blot assay, whereas FCS clearly indicated different cytokines, for example, growth factors.
In vitro wound models are necessary to quantitatively characterize the molecular mechanisms of wound healing and cell migration. Several models have been developed for delineation of cellular repair.34,35 The wounding of epithelial cells in monolayers is an adequate method to study migration and proliferation of cells into wounded areas.36,37 In our wound model, there was no difference between WF and EM. This could be explained by the existence of almost the same cytokines and growth factors in FCS and WF, but in different concentrations.
One of the aims of tissue engineering is to use MSCs to cure damaged tissues. Since stem cells are in direct contact with WF both during and after implantation, it is, therefore, important to investigate whether WF has any negative influence on the differentiation potential of stem cells.
The immunoregulatory properties of ADSCs are well described in the literature. 38 According to Puissant et al., ADSCs do not generate in vitro alloreactivity of incompatible lymphocytes and they suppress the lymphocyte proliferative response to mitogens and alloantigens. 38 The in vitro cultivation of ADSCs with WF in our study was successful, although they were both from different individuals. The differentiation potential under cultivation with WF remained unchanged. Nevertheless, there was an alteration in the quantity of surface antigens CD105, CD90, and CD73 during cultivation with WF compared with EM. The expression of cells with both surface marker CD90 and CD73 was down regulated. Although the expression of CD90 alone was enhanced, the expression of CD105 was attenuated. Cytokines released during the wound-healing phase might be responsible for this alteration. They may have an influence on stem cells, thus resulting in improved wound healing. For example, CD90 thymocyte differentiation antigen-1 (Thy-1) is involved in inflammation and wound healing through different mechanisms such as the synthesizing and releasing of growth factors or cytokines. It is also involved in cell–matrix and cell–cell adhesion. 39 The increase in Thy-1 surface markers in ADSC cultivation with WF could be explained by considering the mechanisms of Thy-1 involvement in wound healing and fibrosis.
Based on our observations, WF and its cytokines play an important role in tissue regeneration by activation of cell migration and enhancing cell proliferation. Although there are important issues regarding WF concerning its cytokines and growth factors, there are also potential risk factors for infection. To avoid infections, careful wound management and adequate wound drainage are required. 40 Stem cells are frequently used in tissue regeneration, and the availability of a sufficient cell number is necessary for tissue repair. Stem cells are typically cultivated in media containing FCS, which is xenogenic, and may cause an antigen reaction or viral infection. Kakudo et al. showed enhancement of ADSC proliferation treated with platelet-rich plasma. 24
In conclusion, in vitro expansion of stem cells with WF proved possible. The proliferation and migration effect of WF on stem cells was significantly better compared with FCS. WF and its cytokines could represent one potential explanation for the migration of stem cell toward the wound. Future investigation is warranted to clarify the significance of the shift of surface markers.
Footnotes
Disclosure Statement
No competing financial interests exist.