
Abstract
Nano- and microscale topographical cues play critical roles in the induction and maintenance of various cellular functions, including morphology, adhesion, gene regulation, and communication. Recent studies indicate that structure and function at the heart tissue level is exquisitely sensitive to mechanical cues at the nano-scale as well as at the microscale level. Although fabrication methods exist for generating topographical features for cell culture, current techniques, especially those with nanoscale resolution, are typically complex, prohibitively expensive, and not accessible to most biology laboratories. Here, we present a tunable culture platform comprised of biomimetic wrinkles that simulate the heart's complex anisotropic and multiscale architecture for facile and robust cardiac cell alignment. We demonstrate the cellular and subcellular alignment of both neonatal mouse cardiomyocytes as well as those derived from human embryonic stem cells. By mimicking the fibrillar network of the extracellular matrix, this system enables monitoring of protein localization in real time and therefore the high-resolution study of phenotypic and physiologic responses to in-vivo like topographical cues.
Introduction
The heart in particular has a complex architecture that spans multiple length scales, from the nano- to the microscale. Comprised of layers of myocardial and matrix fibers, its anisotropic architecture helps coordinate mechanical contraction as well as electrical propagation.15,19 However, commonly used cardiomyocyte (CM) culture and tissue engineering methods do not provide the proper environmental factors to allow CM to respond morphologically, mechanically, or physiologically as they do in the native tissue.15,20 Fabrication of cell culture substrates that mimic the native environment found in the heart may improve the culture conditions of cells for the development of functional cardiac tissue.21–24
The alignment of CM has been studied for the last decade using different microfabrication (e.g., microcontact printing, abrasion, photolithography, hot embossing, electrospinning, and laser ablation) approaches.25–30 Studies have also shown that these microtopographic cues have a greater effect on cell alignment than electrical cues. 25 However, while most previous studies examined the effects of mechanical cues at the 10s to 100s of microns, recent studies indicate that structure and function at the heart tissue level is exquisitely sensitive to mechanical cues at the nano-scale level as well. Cues at the nano-scale stipulate cell mechanics, protein expression, and anisotropic action potential propagation. 1 Indeed, the supporting myocardial extracellular matrix (ECM) is composed of aligned fibrils on the order of 100 nm in diameter with considerable variations in distances between the fibrals. 15 However, most current nanofabrication approaches are complex and expensive (e-beam lithography and nanoimprint lithography) and therefore inaccessible to most biological studies. While less expensive alternative approaches are available, there is a significant trade-off; methods such as colloidal lithography have randomly ordered and oriented features and are therefore inappropriate for anisotropic alignment studies. 14 Moreover, these systems do not span the inherent multiscale structures of the native heart.
To address this chasm, we are introducing a tunable, ultra-rapid, robust, and inexpensive nonphotolithographic fabrication method to create cell culture substrates with controllable nano- and microscale cues. The alignment grooves were created by leveraging the mismatch in stiffness between a prestressed polymer sheet and an overlying thin metal film.31,32 When the plastic sheet retracts upon heating, the stiffer metal film buckles in a controllable manner causing wrinkles. Using this unique self-assembled multiscale topographical substrate, we then studied CM alignment. We created a bio-mimetic fibrillar ECM for the CM culture by first coating our polydimethylsiloxane (PDMS) substrate with fibronectin and laminin. We then studied the response of the subcellular architectural rearrangement by fluorescently staining nuclei, actin, cardiac troponin (CTN-I), gap junction protein connexin-43 (Cx43), as well as the fascia adheren protein, N-cadherin, and the focal adhesion protein, vinculin. We quantified the alignment and cell-tissue morphology from 2 to 7 days and show that the cells robustly align upon attachment (within 2 days). This work demonstrates that we can align both murine neonatal CM (NNCM) and CM derived from human embryonic stem cells (hESC) in vitro using our biomimetic surface topography. Importantly, the CM derived from hESC in vitro are capable of sensing and responding to appropriate nano- to microscale mechanical cues.
Materials and Methods
Multiscale topography
Metal wrinkles were fabricated as previously reported and described in detail. 31 Briefly, gold–palladium was deposited by sputter coating (SEM Sputter Coater; Polaron) at various thicknesses. We showed that we were able to achieve controllable heterogeneous wrinkle length-scales based on varying thickness of metal coating on prestressed polystyrene (PS) sheets (Grafix); coating thicknesses ranged from 15 to 90 nm, with all thicknesses generating wrinkles ranging from 20 nm to 10 μm and average wrinkles thicknesses ranging from 800 nm to 1 μm and increasing proportionally with coating thickness. 31 Due to the heterogeneous range of thicknesses, all wrinkles tested in preliminary cell alignment studies using stromal cells were able to successfully induce alignment of the cells (data not shown). We chose a 60-nm coating thickness for our studies. After deposition, PS sheets were induced to thermally shrink uniaxially by constraining them from opposite sides (2 inch binder clips; OfficeMax) and heating to 150°C–160°C. The deposited metal layer on top of the PS sheet generated aligned wrinkles serve as a soft lithography mold for generating PDMS microchips for culturing the cells (Fig. 1). The anisotropy and length scale of the wrinkles were determined by performing a Fast Fourier transform of the scanning electron micrograph (Fig. 1); the critical length scale was plotted as a probability function and range in critical dimension from the 100s of nm to several microns. Similarly, the depth of the wrinkles, as determined by profilometry (Tencor Alpha-Step 200), was on the same range with self-similar features from the 100s of nm to ∼3 μm.
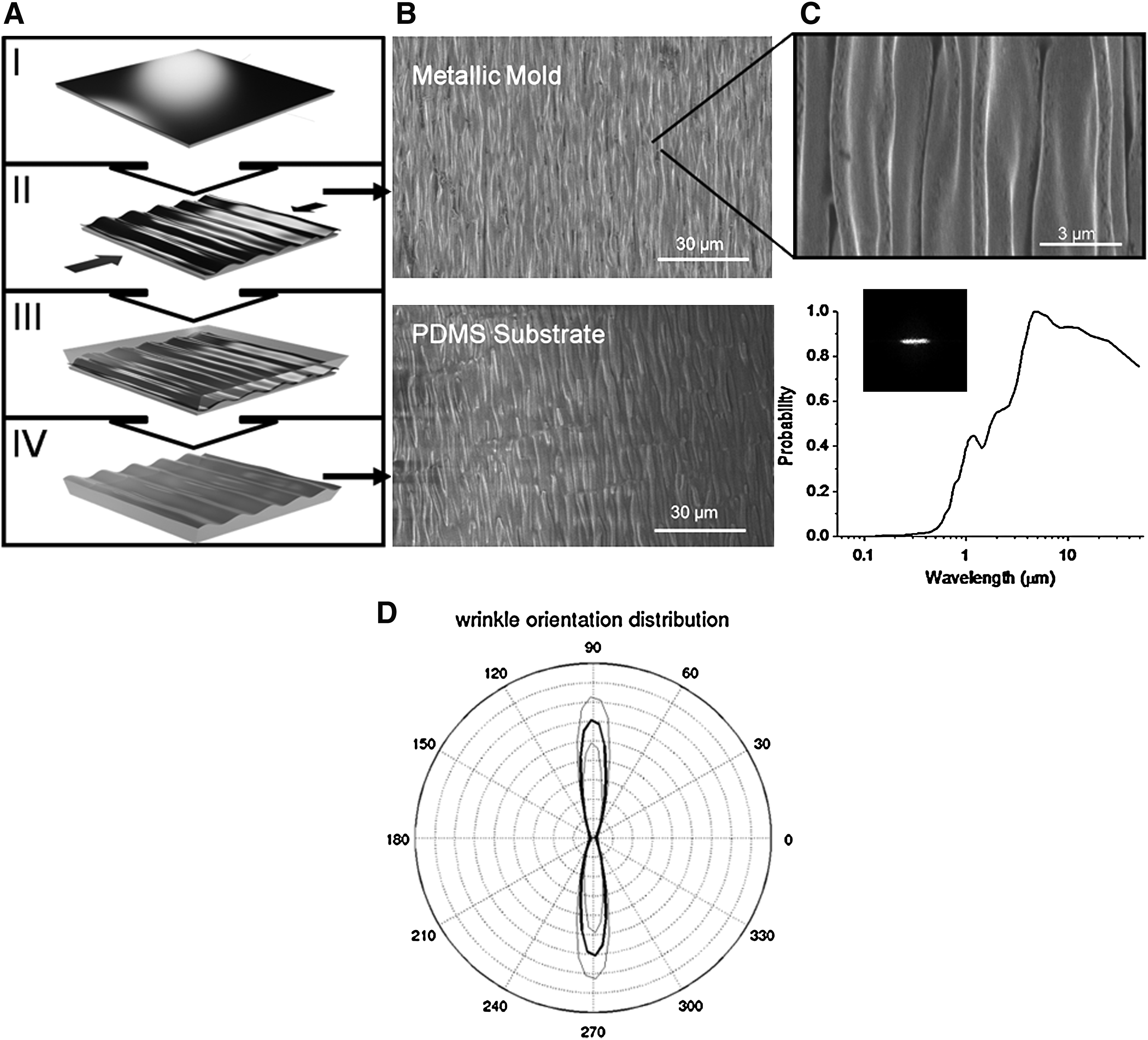
Fabrication and characterization of multiscale wrinkle substrate.
A mixture of 10:1 ratio of PDMS and curing agent (Sylgard 184 Silicon Elastomer Kit; Dow Corning) was poured on the metal mold and set to cure at 75°C. It was then peeled off and cut into to a circle with diameter 15 mm to fit into 24-well plates. Controls with flat PDMS were performed following the same procedure. The chips were sterilized following standard procedures and then coated with 25 mL of 1:1 1 mg/mL laminin (Invitrogen) and 1 mg/mL bovine fibronectin (Sigma Aldrich). As apparent from Figure 1C, the features are highly anisotropic and the self-consistent structures span from the 400 nm to the 50 μm mimicking the fibrillar network of the heart's ECM. Because the metal integrates into the plastic when the plastic retracts, the metal wrinkles are robust and can withstand ultra-sonic fatigue testing as well as multiple moldings without delamination. Moreover, the ease and simplicity of this microfabricaiton design for generating the wrinkle template molds can easily be replicated in any biological laboratory with only minimal equipment; a sputter coating system and an oven to thermally shrink the PS sheets.
Murine NNCM isolation
All animal procedures were reviewed and approved by the UC, Merced Institutional Animal Care and Use Committee (IACUC). C57BL/6 (B6) mice were purchased from Jackson Laboratories or bred in house and housed in sterile microisolator cages. Neonatal mice were euthanized after birth by hypothermal shock. Cell extraction was performed as previously described with some modifications.21,25,27 Briefly, hearts were extracted from neonatal mice and placed in chilled 1× PBS (calcium magnesium free). Blood was removed by squeezing the ventricles, rinsed with PBS, and placed in HBSS for 10 min. Auricles were removed, ventricles quarter minced, and incubated with 0.05% trypsin–EDTA (Gibco) in HBSS for additional 10 min at 37°C. The trypsin solution was removed and the predigested tissue transferred to 0.2% collagenase-type 2 (Worthington, USA 220 units/mL) in HBSS, vortexed for 1 min, incubated in water bath at 37°C for 3 min, vortexed for 1 min again, and set to sediment for 1 min. Liquid solution was transferred to a new falcon tube containing DMEM/F:-12 (Hyclone Thermoscientific) enriched with 20% fetal bovine serum (Atlanta Biologicals) to stop enzymatic digestion. Two cycles were repeated to completely digest the tissues. Cell suspension were filtered using a 70-micrometer nylon mesh (BD Bioscience) followed by centrifugation for 15 min at 2500 rpm and 4°C and then resuspended in the culture medium. CM were enriched by the following differential attachment technique. Cells were incubated in a T75 flask for 1 h. Then, nonadherent cells were collected and centrifuged for 10 min at 2500 rpm at 4°C. Cells were counted by trypan blue exclusion test. The enriched CM were then seeded onto PDMS substrates at a concentration of 7.5×105 cells per chip, about 1.80 cm2 in 1 mL of media containing 20% fetal bovine serum, 5% horse serum (Invitrogen), 100 U/mL penicillin–streptomycin (Gibco), 2 nM L-Glutamine (Gibco), 3 μM sodium pyruvate (Cellgro Mediatech), 1 μg/mL bovine insulin (Sigma), and 0.1 mM nonessential amino acids (Gibco) at standard conditions 95% air and 5% CO2 at 37°C. The FBS in the medium was reduced to 10% to prevent overgrowth of cardiac fibroblast or endothelial cells.
Purification of cardiac fibroblasts
Additionally, the nenonatal mouse cardiac fibroblasts (CF) were extracted and purified to analyze their behavior on the wrinkled substrate and their potential role in guiding the alignment of the NNCMs. For isolation of CF, the same protocol for cardiomyocyte extraction was performed except that the adherent cells were collected and cultured rather than the nonadherent cells. After 24 h in media containing DMEM (Gibco), 20% FBS, and 100 U/mL penicillin–streptomycin, the CF were enriched further by the selective adhesion method described above. Selective adhesion was then repeated three times to obtain highly enriched fibroblast cultures.
Immunostaining
Cells were washed with PBS, fixed with 4% paraformaldehyde, and permeabilized with 0.1% Triton X-100 with 1% BSA. Cells were then washed again and blocked with 10% BSA and normal goat serum (Jackson Immunoresearch Laboratories). Cells were incubated for 30 min with anticardiac troponin I (CTN-I; Santa Cruz Biotechnology) to identify the cardiac cells followed by anti-rabbit Alexa 488 (Invitrogen). Cells were then rinsed and costained with rhodamine-phalloidin (Invitrogen). CD31-FITC (eBioscience) was used for the identification of endothelial cells, and CD 90.2-FITC (Thy 1.2; eBioscience) was used for staining the fibroblasts.33,34 Although CD90.2 is not a specific marker of cardiac fibroblasts, it is expressed by these cells in the heart and used to identify fibroblasts by flow cytometry and immunofluorescence analysis. 35 The nuclear staining was performed using DAPI for all immunofluorescence assays.
The CF were characterized by immunofluorescence analysis using antibodies against CD90.2-FITC (eBioscience) and DDR-2 (Santa Cruz Biotechnologies) followed by anti-rabbit Alexa-488 (Invitrogen), a marker specific for cardiac fibroblasts, followed by actin staining using rhodamine-phalloidin (Sigma) to analyze fibroblast morphology as previously described. 36 Anticardiac troponin I (CTN-I; Santa Cruz Biotechnology) and anti-VE-cadherin-PE (eBioscience) were used to confirm the absence of cardiomyocytes and endothelial cells in the CF cultures.
For gap junction and fascia adherens protein localization, the cells were fixed with 4% paraformaldehyde, blocked with 1% BSA, and incubated with either connexin-43 (Santa Cruz Biotechnology) or N-cadherin (Abcam), respectively, followed by anti-rabbit Alexa-488 (Invitrogen). For visual localization of the focal adhesions, an antibody against vinculin (Santa Cruz Technology) was used followed by Alexa-488. Samples were mounted on a cover glass using mounting medium (Vector Laboratories) and imaged with an inverted fluorescent microscope (Nikon Eclipse TE2000-U) and digital camera (Photometrics Coolsnap).
To characterize myofilament alignment in the hESC-CM, the cells were fixed with 4% paraformaldehyde followed by permeabilization with 0.2% Triton. After blocking with 10% goat serum and 1% BSA, the cells were labeled with mouse monoclonal tropomyosin antibody (T9283; Sigma) and then Alexa Fluor 488 goat anti-mouse IgG1 (Invitrogen). Hoechst 33342 was used as the nuclear counterstain.
Flow cytometry analysis
For CM population analysis, the cells were washed with PBS, fixed with 4% paraformadehyde for 15 min, and permeabilized with 0.1% Triton X for 10 min. The cells were then rinsed with PBS, centrifuged at 2500 rpm for 5 min at 4°C, and blocked with 10% BSA and normal goat serum. The cells were stained with anti-CD31-FITC (eBioscience) for endothelial analysis, anti-CD90.2-FITC (eBioscience) for fibroblast analysis, and anti-Cardiac Troponin I (CTN-I; Santa Cruz Biotechnology) followed by FITC-conjugated donkey anti-rabbit (Biolegend) for quantification of the cardiac cells. Analysis was performed with a flow cytometer BD-LSRII (Biosciences) by acquiring 1 million cells per run, and analyzed using FlowJo 7.5 software. The CM, endothelial, and fibroblasts compositions were measured for three separate neonatal heart isolations (n=3) consisting of 4–7 neonatal hearts per isolation.
hESC culture and cardiac differentiation
The hESC (WiCells) HES2 line (NIH code: ES02) was cultured under feeder-free conditions on hESC-qualified Matrigel (Becton Dickinson) in mTeSR (Stem Cell Technologies) and differentiated as described by Yang et al. 37 Briefly, hESCs were detached using trypsin (Invitrogen) and transferred to low-attachment plates to allow aggregate formation. The aggregates were cultured in suspension for 12 days in StemPro-34 medium (Invitrogen) with 2 mM L-glutamine (Invitrogen), 150 μg/mL Holo-Transferrin (Sigma), 0.45 mM monothioglyerol (Sigma), and 50 μg/mL ascorbic acid (Sigma) with addition of 10 ng/mL BMP4 (R&D Systems), 3 ng/mL activin A (R&D Systems), and 5 ng/mL bFGF (Invitrogen) from day 1–4, 10 ng/mL VEGF (R&D Systems), and 150 ng/mL DKK1 (R&D Systems) from day 4–8, followed by 10 ng/mL VEGF, 150 ng/mL DKK1, and 5 ng/mL bFGF from day 8–12. This directed differentiation protocol yielded >50% of spontaneously beating cardiospheres.
To isolate the hESC-CM, the spontaneously contracting cardiospheres were picked and digested into single cells with collagenase II (1 mg/mL) at 37°C for 30 min, followed by 0.05% trypsin–EDTA (Invitrogen) at 37°C for 5 min. The isolated cells were then cultured in KB solution containing (mM): 85 KCl, 30 K2HPO4, 5 MgSO4, 1 EGTA, 2 Na2-ATP, 5 pyruvic acid, 5 creatine, 20 taurine, and 20 D-glucose, at room temperature for 30 min, followed by plating onto the wrinkles. Because these methods generated low numbers of purified CMs, we also proceeded to transduce these hESCs with the MLC2v promoter that drives both GFP fluorescence and zeocin antibiotic resistance to more easily indentify and purify the ventricular CMs.
To obtain GFP-labeled ventricular cardiomyocytes (GFP-VCM), single cells isolated from cardiospheres were plated and cultured with the medium containing 80% DMEM, 20% FBS defined (HyClone), 1 mmol/l L-glutamine, 1% NEAA, and subsequently transduced with recombinant LV-MLC2v-GFP particles at multiplicity of infection (MOI) of 5. The MLC2v promoter simultaneously drove the expression of GFP as well as zeocin for antibiotic resistance in the transduced hESC-VCM. The GFP-VCM were then purified with a 5-day treatment of zeocin (300 μg/mL).
Image analysis
The images were processed further to quantify the distribution of orientations using custom scripts written in MATLAB (MathWorks, Inc.). Images of labeled actin, CTN-I, Cx43, and N-cadherin were filtered to estimate the image gradient at each pixel location using a Guassian derivative (σ=2). The distributions of the orientations were estimated by computing a histogram of gradient orientations, where the contribution of each pixel is weighted by the gradient magnitude. This weighting limits the contribution of pixels in low contrast regions of the image where gradient orientation estimates are uncertain. The orientation histogram was computed for n=12 disjointed 300×300 pixel subwindows in each image, and the standard deviation was computed across the subwindows.
An additional measure of cell alignment was also computed by detecting DAPI labeled nuclei and estimating the orientation of the major axis of each nucleus. Nuclei were segmented by thresholding followed by morphological processing and watershed segmentation to separate overlapping nuclei. The orientation of each segmented nucleus was estimated by computing the moment matrix of the segment and identifying the major and minor axes. Potential elongation of the nuclei was computed as the ratio of the lengths of the major and minor axes.
Results and Discussion
Cell population assessment
We initially used NNCM for our studies because they are relatively immature cells and can be cultured more easily in vitro, compared to adult CM that undergo profound morphological and physiological changes in long term culture. 38 Unfortunately, neonatal mouse hearts are limited by their size and thus in the number of cells available for experiments. Isolation of neonatal ventricular CM is challenging, in part because these cells are fully differentiated and cannot be expanded in vitro. 38
We hypothesized that the inclusion of noncardiac cell populations would benefit our CM cultures by inducing them to physiologically respond more like native tissue. We consistently isolated ∼1 million cells per neonatal mouse heart by following the general protocol described by Heidi et al. 27 The heterogeneous cellular composition of the heart includes CM, endothelial cells, fibroblasts, and smooth muscle cells.39,40 In a previous study, it was also shown that the lack of cardiac fibroblasts in CM culture systems negatively affect the alignment, elongation, and network formation of CM. 41 It has also been shown that the non-CM accessory cell populations are advantageous in establishing viable in vitro cultures of CM by aiding in the secretion and degradation of ECM elements that promote CM survival. 39 Further, increased apoptosis of CM was observed in cultures with only CM. Additionally, endothelial cells promote CM survival and enhance spatial organization in a 3-dimensional configuration by establishing CM-endothelium interactions. 42 These previous studies reveal a value in including some accessory cells in the CM cultures; therefore, we did not attempt to completely purify the CM in our cultures.
However, because fibroblasts and endothelial cells are highly proliferative, especially compared with CM, it is also helpful to maintain these accessory populations at lower levels in the cultures. 43 We used a differential attachment plating technique to enrich our CM populations to reduce the number of fibroblasts and endothelial cells in our cultures. We then quantified the percentage of the enriched cardiac cell populations using CTN-I, a specific marker for CM, by flow cytometry analysis. 37 Based on this marker we were able to identify an average (n=3) of at least 70% CM purity in our sorted population with <5% fibroblast and <10% endothelial cells (Fig. 2A). To maintain these accessory cell populations at a low proliferative rate, we reduced the concentration of serum in our media from 20% to 10%.

Characterization of the purified neonatal cardiac cells.
We also examined the purified cardiac fibroblast (CF) from our mixed populations of cells isolated from the neonatal mouse heart to analyze the response of these cells on the wrinkled substrate and potential role in guiding the alignment of the NNCMs. The purity of the CF was verified based on CD90.2 (Fig. 2B) and DDR-2 (Fig. 2C) staining with no endothelial cells observed based on VE-cadherin (Fig. 2D) staining. Interestingly, the purified CF do not align with the wrinkles (Fig. 2F), and appear to fill into the spaces between the cardiac cells (Fig. 2G). This analysis additionally indicates that the CF are not inducing alignment of the CM cells on the wrinkled topology.
Fibrillar ECM
The incorporation of ECM proteins in the CM culture environment is thought to be important for facilitating appropriate CM-ECM adhesions.44,45 Therefore, we performed experiments to determine CM attachments to different ECM proteins by coating our substrates with fibronectin, collagen I, and laminin and combinations of these ECM proteins. We observed that the combination of laminin and fibronectin induced more cells to attach to the substrate (data not shown). Hence, corona treatment before ECM coating and combination of fibronectin and laminin facilitate cell attachment and the generation of confluent CM monolayers. These findings are similar to previous studies where it has been shown that laminin and fibronectin induced more CM to attach compared with collagen I or IV, and maintained cell attachment from day 1 through day 8 of culture.
46
Using immunofluorescent analysis, we observed the distribution of laminin and fibronectin coatings on the wrinkled surface (Supplementary Fig. S1B; Supplementary Data are available online at
Cell alignment
We examined CM alignment at cellular and subcellular resolution induced by our multiscale guidance topography over a period of 7 days. For alignment experiments, the cells were cultured in three different chips and repeated three times to ensure consistent results. The actin filaments were stained to observe the internal architecture of cells. To distinguish CM from the other cell types, CM were also stained for CTN-I. We observed that the cells began plating down on both substrates within the first 24 h (Fig. 3A). After 48 h most of the cells attached and covered most of the surface (Fig. 3C). The cells also remained robustly aligned and a confluent connected layer is observed by day 7 (Fig. 3G). In controls, cardiac cell cultures were also more confluent, but not aligned (Fig. 3H). We expect that the saturated cell population at day 7 is due to the proliferation of noncardiac cells. Image analysis (Fig. 3I) shows the distinct alignment condition versus the control. However, because most of the cells robustly align upon attachment, temporal information could not be obtained from these time points.

Alignment of neonatal cardiac cells on wrinkles.
The video (SV1) demonstrates that the cells can be kept aligned and healthy for extended periods (video taken at day 14). The beating of the aligned cells was more coherent and synchronous compared with the cells cultured on the control flat substrate (SV2). In agreement with previous studies, the cells attached more robustly to the nano-topography and did not detach as in the case of the flat surface. 47
Through actin/CTN-I staining, we also observed the interaction between CM and noncardiomyocyte cells. Recent studies indicate that heart cell elongation and alignment was enhanced by cocultures with cardiac fibroblasts (CF) via matrix metalloprotease (MMP)-dependent mechanisms. 41 Our biomimetic cell culture platform captures both the effects from the indigenous fibroblasts and endothelial cells as well as native topography. CNT-I positive cells appeared to connect and form a defined and consistent network with each other, whereas negative CTN-I accessory cells fill the spaces (Fig. 2G).
To further quantify alignment of cells with the substrate, we examined nuclei alignment. Nuclei tend to be elongated with the direction of the alignment of the actin filaments as previously described. 48 Using image analysis, we automatically segmented the nuclei shapes (Fig. 4A). We measured that the nuclei elongated by a factor of 1.43 (ratio of major to minor principal axis). A two-sampled t-test shows that the elongations of the nuclei were the same in the control and wrinkled substrates (p>0.12). However, the distribution of orientations of the principal axis was substantially different in the aligned cells. Figure 4B shows the distribution of nuclei orientations in control and wrinkled substrates. On the wrinkled substrate, 54% of cell nuclei were aligned within ±20° of the substrate.
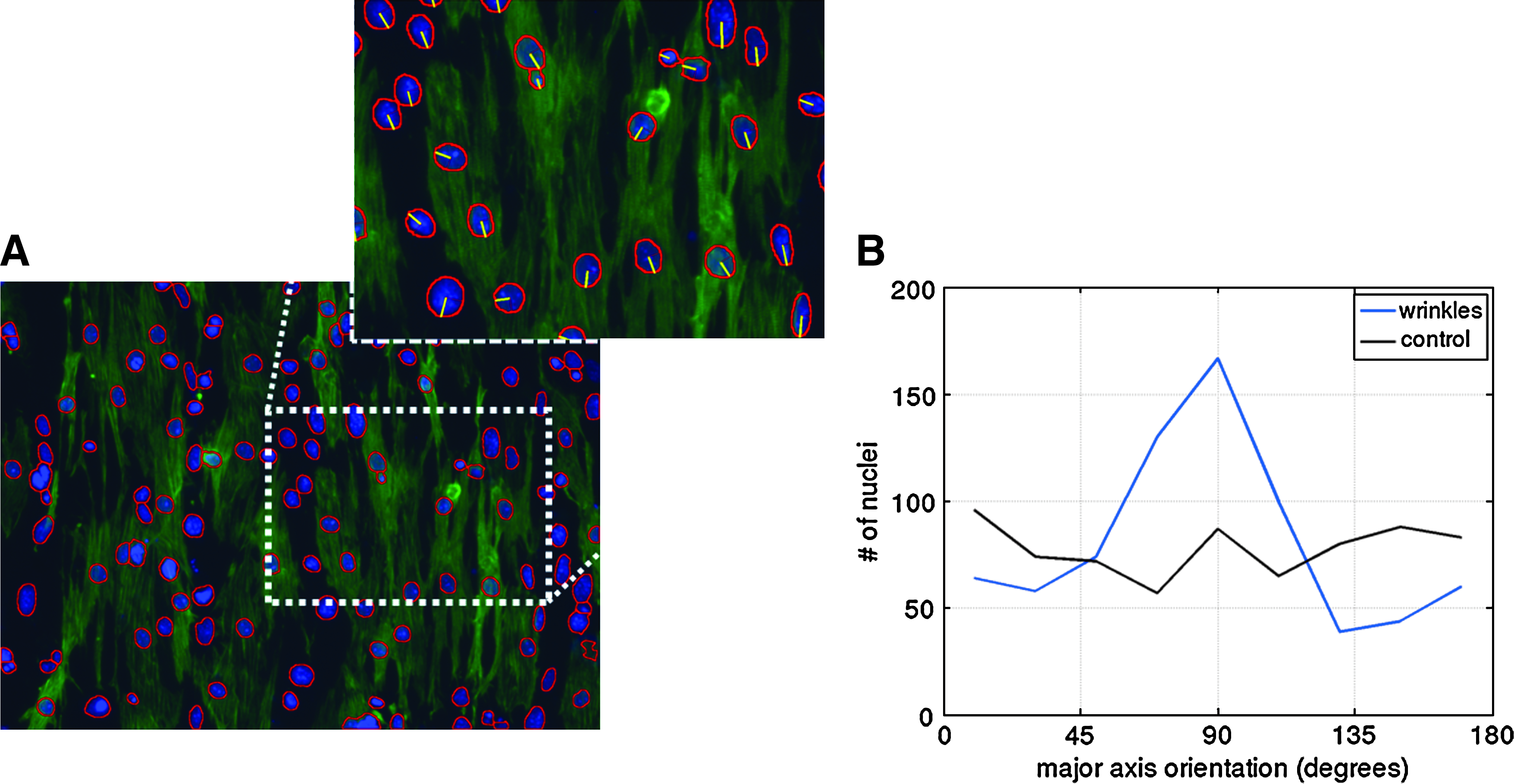
Nuclei alignment on wrinkles. Image processing was used to detect the DAPI-labeled nuclei.
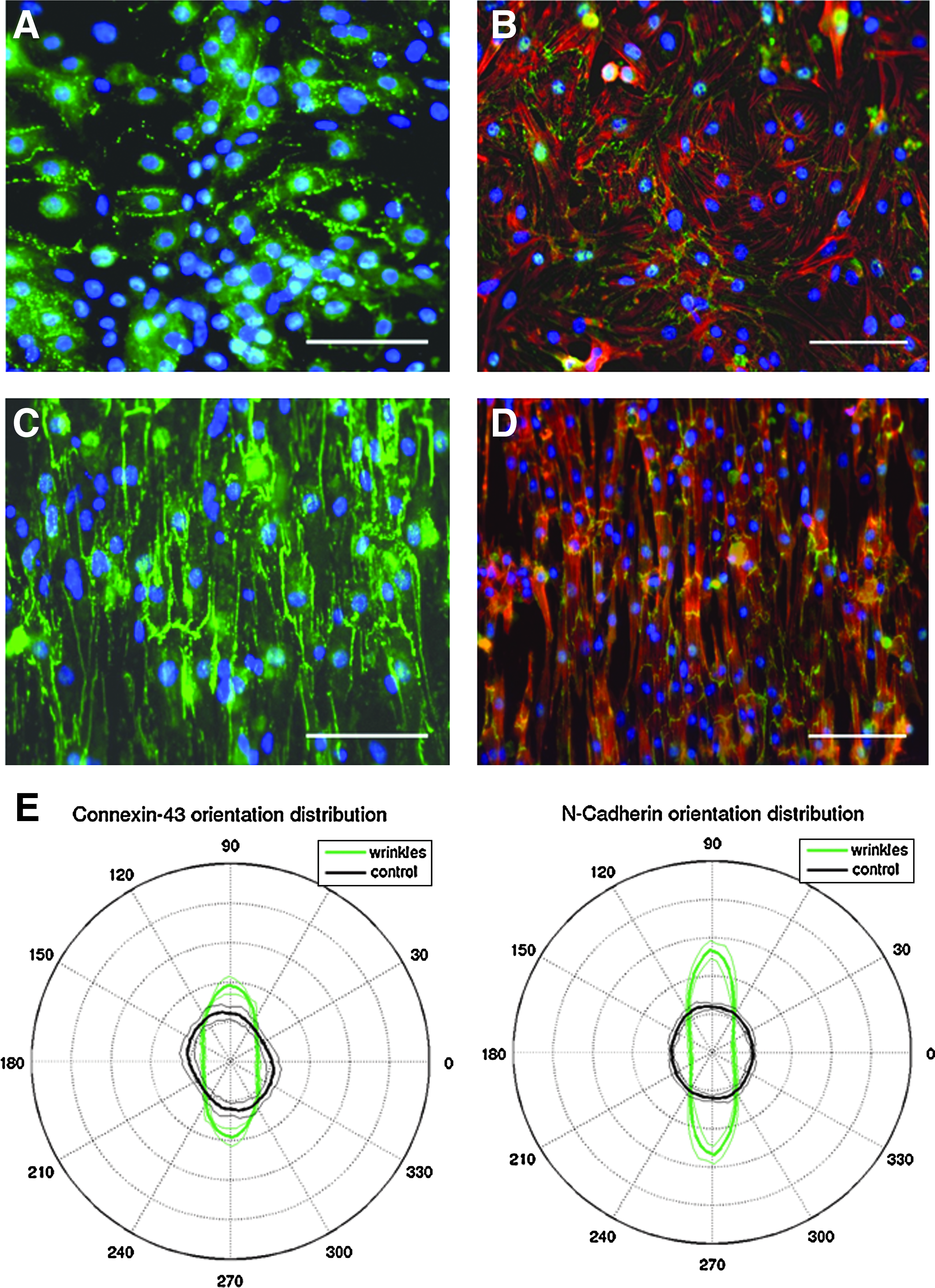
To confirm our hypothesis that the aligned cells connect and form a defined and consistent network with each other, we stained the cells for gap junction protein Cx 43 (Fig. 5A, C), the fascia adheren protein N-cadherin (Fig. 5B, D), as well as the focal adhesion protein vinculin (Supplementary Fig. S2). 5 As expected, the proteins were better localized at the cell–cell junctions on the aligned cells as compared to the peripherally distributed expression on the isotropic cells. We also analyzed the localization and the anisotropic orientation distribution of Cx-43 and N-cadherin (Fig. 5E) and quantitatively show alignment based on the expression patterns of these proteins on the wrinkles compared with the flat substrate.
Cardiac-like tissue from aligned CMs. Fluorescent micrographs of cardiac cells cultured on
We next attempted to align hESC-CMs. The control hESC-CMs cultured on flat substrates were randomly oriented cells with no obvious organization (Fig. 6A–C, top images). Conversely, after attaching to the wrinkled substrate (Fig. 6A–C, bottom images), the hESC-CMs aligned and also displayed the typical banding pattern consistent with organized sarcomeric structure patterns. We also analyzed the orientation based on nuclei shape (Fig. 6D) and actin organization (Fig. 6E) and quantitatively show that the hESC-CMs exhibit increased alignment on the wrinkled substrates compared with the controls.
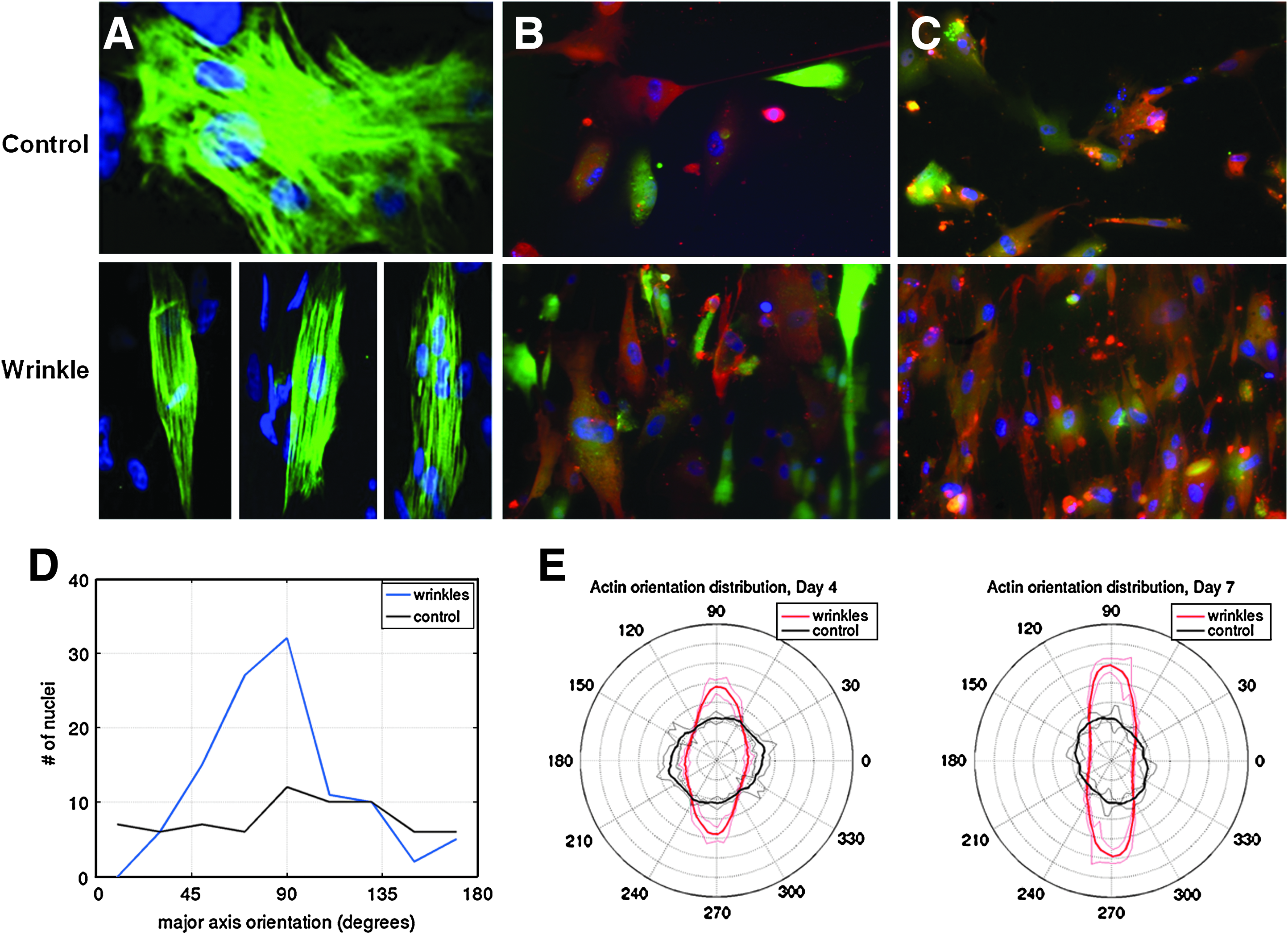
Confocal micrographs of hESC-derived CMs alignment on wrinkles.
In conclusion, here we have presented an easy, inexpensive, and rapid method for the fabrication of nano- to microtopographies to induce and study the cell alignment process by contact guidance, offering insight into the cell alignment process as well as in the formation of cardiac tissue-like constructs. Integration of robust and reproducible biomimetic surfaces into CM culture systems that controllably induce cell orientation may be useful for electrophysiological and pharmacological studies. Moreover, it may lead to the development of tools for tissue engineering such as patterned substrates to generate aligned tissue grafts for heart repair. The incorporation of cardiac cell populations such as CM, cardiac fibroblasts, and endothelial cells enables the reformation of aligned cardiac tissue monolayers. We demonstrated that this platform can also align CM derived from hESC, and that this alignment aids in the development of appropriate internal organization needed for cellular contraction. Our substrate could therefore be used as a tissue engineering tool for the generation of robustly aligned cardiac cell sheets using cells derived from hESC for heart injury repair.
Footnotes
Acknowledgments
Funding for this project was supported by the California Institute of Regenerative Medicine (CIRM) and Shrink Nanotechnologies. We would also like to thank UC Irvine's INRF facilities and Chi-cheng Fu.
Disclosure Statement
The author, Michelle Khine, is the scientific founder of Shrink Nanotechnologies, but receives no compensation nor does she have any financial interest in the company. Terms of this arrangement have been reviewed and approved by UC Irvine in accordance with its conflict of interest policies.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.