
Abstract
In light of the increasing need for differentiated primary cells for cell therapy and the rapid dedifferentiation occurring during standard in vitro cultivation techniques, there is an urgent need for developing three-dimensional in vitro systems in which expanded cells display in vivo-like differentiated phenotypes. It is becoming clear that the natural microenvironment provides the optimal conditions for achieving this aim. To this end, we prepared natural decellularized scaffolds of microscopic dimensions that would allow appropriate diffusion of gases and nutrients to all seeded cells. Scaffolds from either the lung or the liver were analyzed for their ability to support growth and differentiation of progenitor alveolar cells and hepatocytes. We observed that progenitor alveolar cells that have been expanded on plastic culture and thus dedifferentiated grew within the lung-derived scaffolds into highly organized structures and regained differentiation markers classical for type I and type II alveolar cells. The cells generated proper alveolar structures, and only 15%–30% of them secreted surfactant proteins in a localized manner for extended periods. Vice versa, liver-derived scaffolds supported the differentiation state of primary hepatocytes. We further demonstrate that the natural scaffolds are organ specific, that is, only cells derived from the same organ become properly differentiated. A proteomic analysis shows significant different composition of lung and liver scaffolds, for example, decorin, thrombospondin 1, vimentin, and various laminin isoforms are especially enriched in the lung. Altogether, our data demonstrate that complex interactions between the seeded cells and a highly organized, organ-specific stroma are required for proper localized cell differentiation. Thus, our novel in vitro culture system can be used for ex vivo differentiation and organization of expanded primary cells.
Introduction
We have developed a novel 3D cell culture platform that takes the advantage of the natural microenvironment of the organ and thus comprises the right composition and ratio of ECM components and growth factors. This platform is based on our previous reports showing that organ fragments of 300 μm thickness, which maintain the natural organ architecture, remain viable and transcribe tissue-specific genes for long periods both in culture9,10 and when encapsulated and implanted into hosts. 11 We reasoned that such fragments could constitute the basis to prepare cell-free organ-specific microscaffolds. Here, we show that these decellularized scaffolds can be used for growing primary cells without the need for vascularization, because their microscopic thickness allows appropriate diffusion of nutrients and gases. We further demonstrate that lung-derived scaffolds derived in this manner can direct the differentiation of adult alveolar progenitor (AP) cells that have been expanded in vitro on plastic culture plates and can instruct their proper localization and function within the matrix. Likewise, liver-derived scaffolds maintained the differentiation state of primary hepatocytes. Although these microscaffolds cannot generate a whole organ, these natural scaffolds provide the conditions required for in vitro expansion of organ-derived cells and for supporting their differentiation. This system is also ideal for studying ex vivo the intricate interplay between primary cells and their natural microenvironment.
Materials and Methods
Animal experiments were performed under the guidelines and approval of the Animal Care and Use Committee, The Faculty of Science of the Hebrew University, Jerusalem, Israel.
Preparation of lung- and liver-derived fragments
Lungs and livers were taken from Lewis rats (200–300 g), and individual lobes of either lung or liver were washed twice with Dulbecco's modified Eagle's medium (DMEM; Biological Industries, Beit Haemek) and cut transversely into 300-μm-thick fragments using a Sorvall TC-2 tissue chopper. The fragments were then washed 2–3 times with DMEM.
Preparation of lung- and liver-derived microscaffolds
Organ fragments prepared as described earlier were immersed in 0.5% Triton X-100 and 20 mM ammonia solution for 15 min at 37°C, followed by gentle pipetting to mechanically remove the cellular components from the ECM. The matrix preparations were then washed five times with phosphate-buffered saline (PBS) and three times with double distilled water and frozen at −70°C until required. Lung- or liver-derived microscaffolds were thawed and washed three times in PBS and twice in culture media prior to using in culture.
Isolation of AP cells
Lungs were dissected from 4- to 5-week old C57BL/6 mice. The tissue was cut into small pieces and then digested using 0.25% trypsin–0.05% EDTA solution for 30–40 min at 37°C, followed by gentle pipettization to mechanically separate cells. The cells were collected by centrifugation and resuspended in culture medium (DMEM supplemented with 10% fetal calf serum, 100 U/mL penicillin, 0.1 mg/mL streptomycin, and 2 mM
Isolation of hepatocytes
Hepatocytes were isolated by collagenase perfusion of liver from C57BL/6 mice following the method described by Seglen. 13 Briefly, after a two-step perfusion of the whole animal with calcium-free buffer and collagenase, cells were dissociated and collected by centrifugation. The cells were then suspended and cultured with hepatocyte culture medium (Cambrex).
Coculture of cells with microscaffolds
Mouse AP cells or hepatocytes were seeded on rat lung-derived microscaffolds or liver-derived microscaffolds and incubated at 37°C in 5% CO2. The reason for using rat as the source of scaffolds was to ensure that no scaffold contaminants were part of the signal obtained in polymerase chain reaction (PCR) analyses when primers specific to mouse genes were used. Five to seven lung-derived microscaffolds were inserted per well or 2–4 liver-derived microscaffolds added per well in 24-well plates and cultured in 1 mL of culture medium. Cell concentration varied from 2×105 to 106 cells/mL per well. After 24 h, the seeded microscaffolds were transferred to new wells to discard nonadherent cells. The medium was changed every 2 days. Incubation took place at 37°C in 5% CO2.
MTT viability staining of liver microscaffolds seeded with primary hepatocytes
For detection of hepatocytes grown on liver-derived microscaffolds, these were washed in PBS and incubated for 10–20 min in 0.5 mg/mL MTT (3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide; Sigma) dissolved in PBS. Viable hepatocytes are stained purple blue.
Cryostat sections
Microscaffolds were fixed for 15 min in phosphate-buffered saline containing 4% paraformaldehyde, rinsed, and embedded in TissueTek (Sakura, Japan). Eight-micrometer-thick cryosections were prepared and nuclei were stained with 10 μg/mL of 4,6-diamino-2-phenylindole dihydrochloride (DAPI; Sigma) in PBS.
Transmission electron microscopy
Microscaffolds samples were fixed in 1% formaldehyde and 2.5% glutaraldehyde, postfixed in 1% osmium tetroxide, dehydrated, and embedded in Epon using standard techniques. Ultrathin sections (60–80 nm thick) were prepared using an Ultratome III (LKB). The sections were collected on 200-mesh thin bar grids, counterstained with uranyl acetate and lead citrate, and examined using a Philips CM 120 transmission electron microscope.
Reverse transcriptase–polymerase chain reaction
RNA isolation and reverse transcriptase–polymerase chain reaction (RT-PCR) were done as previously described.
10
Primer sets for PCR are summarized in Supplementary Table S1 (Supplementary Data are available online at
Immunohistochemistry
Immunohistochemistry was done using standard techniques on cryosections. Sections were incubated overnight at 4°C with primary rabbit antibodies to presurfactant protein B (Chemicon; diluted 1:800), washed with 0.5% Triton X-100/PBS, and incubated for 2 h with a secondary antibody. Cy3-conjugated goat anti-rabbit antibodies (Jackson ImmunoResearch Labs; diluted 1:800) were used for immunofluoresence, whereas horseradish peroxidase-conjugated goat anti-rabbit antibodies (Jackson ImmunoResearch Labs; diluted 1:1000) were used for immunohistochemistry. Negative controls were prepared by incubating the sections with second antibody only. Washing was repeated and sections were counterstained respectively with 10 μg/mL DAPI (Sigma) or with hematoxylin and washed again. Sections were mounted under coverslips and viewed with conventional fluorescence or light microscopy. The percentage of pro-SBP–positive cells was calculated by counting the number of red-stained cells versus the number of DAPI-positive nuclei, which represents the total cell number. Laminin whole-mount staining was done as follows: lung-derived microscaffolds were fixed for 30 min in 4% paraformaldehyde and further fixed with 50% ethanol, 5% acetic acid, and 4% paraformaldehyde for 2 h. Samples were rinsed with PBS, washed with 0.5% Triton X-100/PBS, and transferred to 4% glycine in 0.5% Triton X-100/PBS for 40 min. The samples were then washed again with 0.5% Triton X-100/PBS followed by 2 h incubation with 5% normal goat serum and 1% BSA in PBS at 37°C. Samples were incubated with a polyclonal rabbit anti-laminin antibody (Sigma) diluted 1:50 with 1% BSA in PBS (blocking solution) at 4°C overnight. Samples were then washed with 0.5% Triton X-100/PBS and incubated with secondary antibody diluted in blocking solution for 2 h at 37°C. Washing was repeated and the samples were counterstained with 10 μg/mL DAPI, followed by three 3-min washes in PBS. Negative controls were prepared by incubating the samples with secondary antibody only. No signal was obtained in any of the negative controls done (data not shown).
Environmental scanning electron microscopy
Environmental scanning electron microscope Quanta 200 (FEI Company) operated in low-vacuum mode (0.6 Torr) at an accelerating voltage of 20 kV was used to analyze freshly prepared microlung scaffolds that were fixed in 4% paraformaldehyde for 30 min and transferred to PBS.
In gel proteolysis and mass spectrometry analysis
Decellularized microscaffolds were homogenized in 9 M urea, 20 mM DTT, and 400 mM ammonium bicarbonate. The proteins were separated on 10% SDS-PAGE. Five gel slices were used for mass spectrometry (MS) analysis. The proteins in the gel were reduced (3 mM DTT, 60°C for 30 min), modified with 12 mM iodoacetamide (room temperature for 30 min), and trypsinized (modified trypsin; Promega) at a 1:10 enzyme-to-substrate ratio. The resulting tryptic peptides were resolved by reverse-phase chromatography on 0.075×200 mm fused silica capillaries (J&W) packed with Reprosil reversed-phase material (Dr. Maisch GmbH). The peptides were eluted with linear 65 min gradients of 5%–45% and 15 min at 95% acetonitrile with 0.1% formic acid in water at flow rates of 0.25 μl/min. Mass spectrometry was performed by an ion-trap mass spectrometer (LTQ, Thermo) in a positive mode using repetitively full MS scan followed by collision induces dissociation of the seven most dominant ion selected from the first MS scan. The mass spectrometry data were clustered and analyzed using the Sequest 3.31 software (J. Eng and J. Yates, University of Washington and Finnigan, San Jose), searching against the rat sections of the NCBI NR database. The estimation of the expression was done by the identification coverage of the proteins. To search for ECM-related proteins, GO annotations were determined using the DAVID Bioinformatics Resources 6.7 (NIAID/NIH;
Results
Characterization of organ-derived microscaffolds
The aim of this study was to develop a 3D in vitro system that would allow for expanded cells to display in vivo-like tissue-specific differentiated functions. We hypothesized that the natural microenvironment would be the appropriate source to achieve this aim. To this end, we attempted to obtain acellular organ-derived microscaffolds from rat lung and liver that preserve, as much as possible, the structure of the original ECM of the organ as well as the ECM-associated growth factors. 14 The tissues were cut into 300-μm-thick pieces as previously described9–10 and subjected to a decellularization process. Several protocols to decellularize the tissues were analyzed and the capacity of the resultant microscaffolds to support proper cell function (see below) was tested in a systematic manner. We found that organ fragments immersed in 0.5% Triton X-100 and 20 mM ammonia solution for 15 min at 37°C, followed by extensive washes in PBS, gave microscaffolds with preserved organ structures. Together with the functional analyses, the peptide composition of the decellularized microscaffolds was analyzed by liquid chromatography combined with tandem mass spectrometry LC MS/MS (see below).
Figure 1A presents a high-energy scanning electron microscopy picture of a lung microscaffold, which shows that the microstructure of the alveoli is preserved. Figure 1B illustrates that the basement membrane, as manifested by laminin staining, appears intact. The morphology of the scaffolds was further examined by transmission electron microscopy (TEM) and compared with the morphology of normal lung tissue (Fig. 1C vs. 1D). The acellular scaffolds revealed the typical architecture of the connective tissue of the alveoli. The thin connective tissue layers of the alveolar septum are clearly seen, which separate the preserved cavities one from the other. The fine structures of collagen fibers and basement membranes were also preserved (Fig. 1E, F).
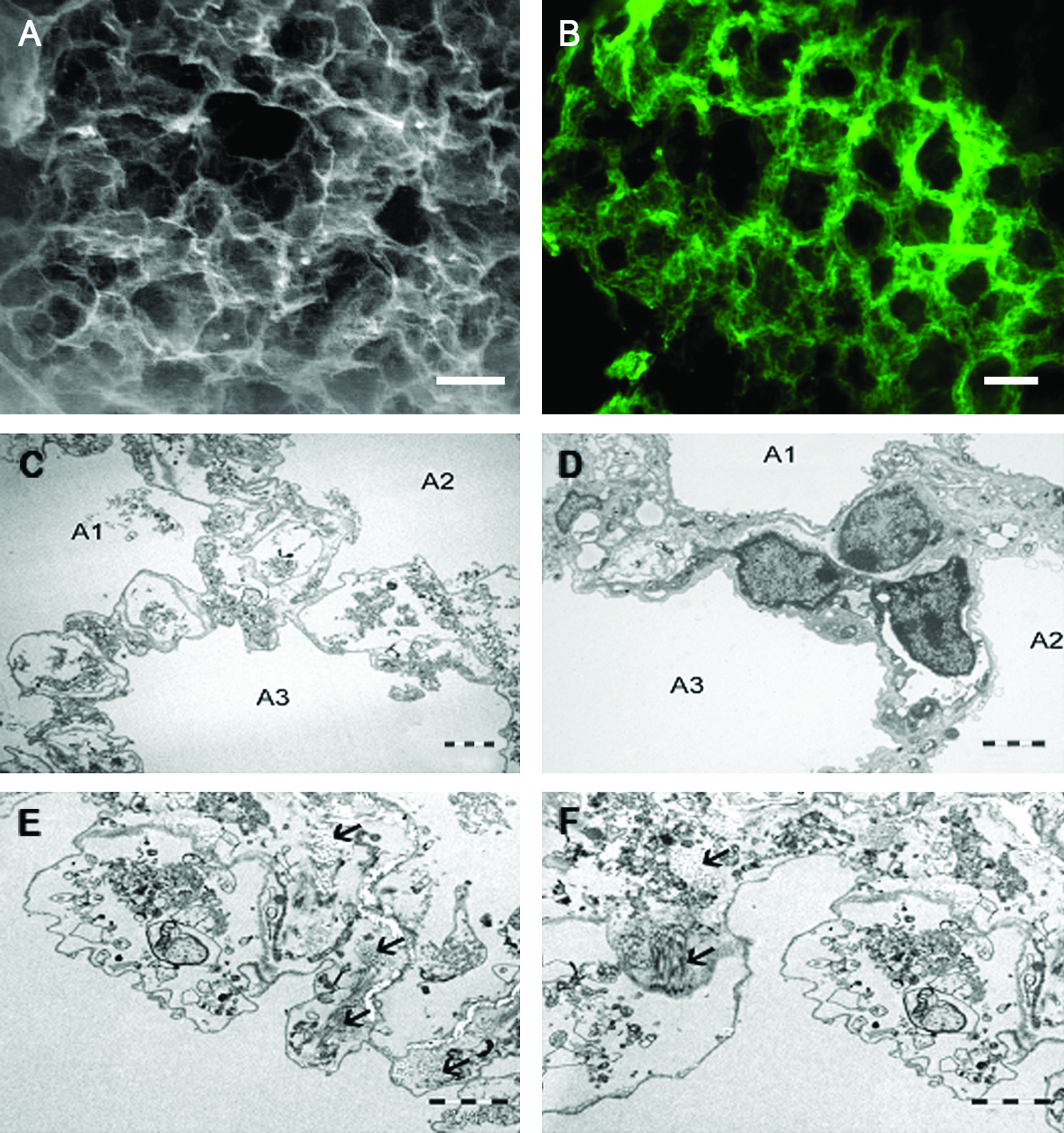
Structure of lung-derived microscaffolds.
Expansion and differentiation of primary hepatocytes on liver microscaffolds
Figure 2A shows that also in liver-derived scaffolds the basement membrane is preserved. Hepatocytes, when seeded on liver-derived microscaffolds, adhered to the scaffolds and remained viable for the whole culture period of 21 days. The hepatocytes did not expand significantly and thus were capable only to partially repopulate the liver-derived microscaffolds (Fig. 2B).
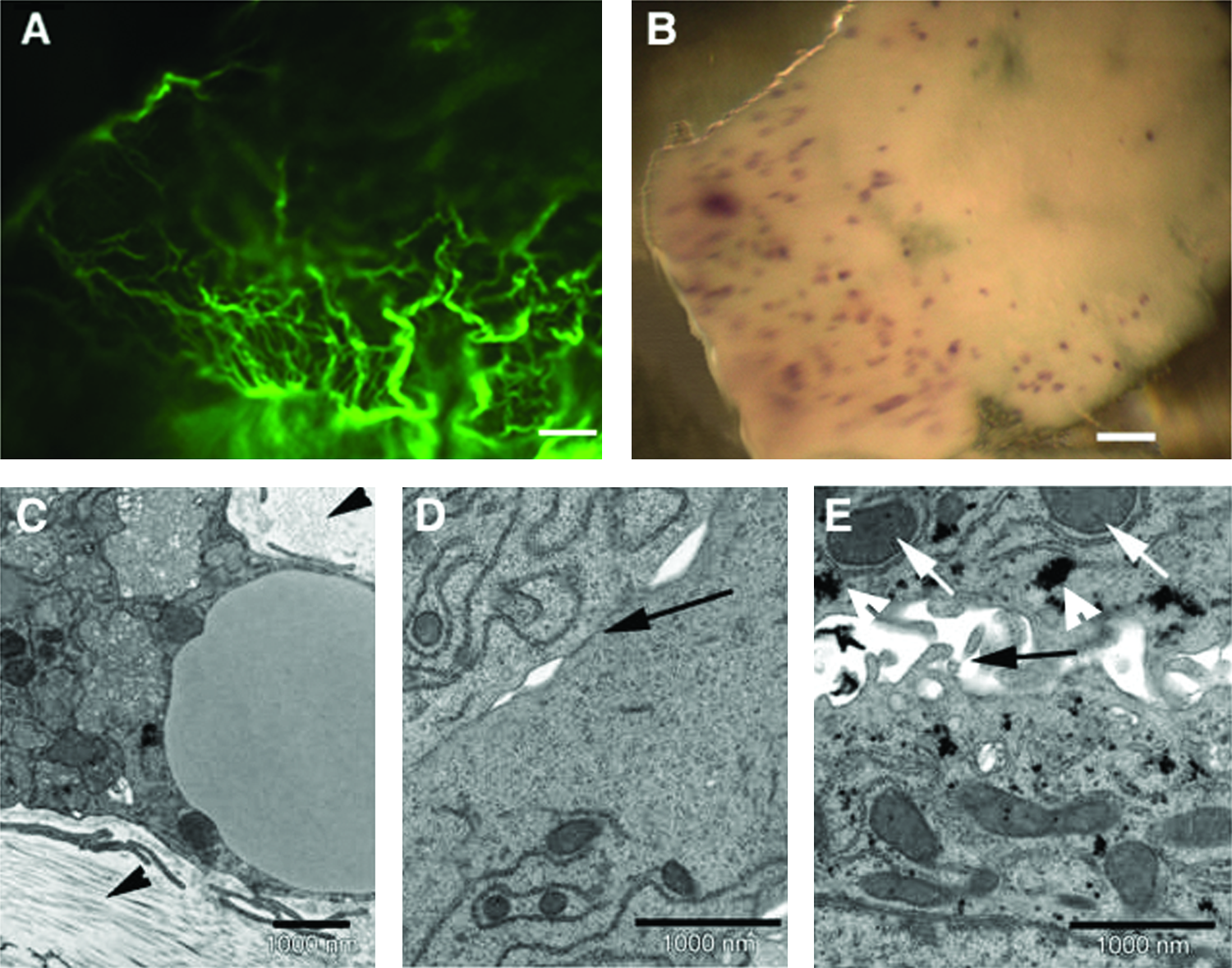
Repopulation of liver microscaffolds with primary hepatocytes.
Despite the limited expansion, hepatocytes formed a well-organized epithelium in these scaffolds after 14 days cultivation with characteristic tight junctions as indicated by TEM analysis (Fig. 2C–E). The hepatocytes, as in the normal liver, were found to present microvilli in a polar fashion (Fig. 2E). Low-density lipoproteins and glycogen aggregates, which are also characteristic of hepatocytes in vivo, were also observed in the liver microscaffolds cultured with hepatocytes (Fig. 2E).
Expansion of adult progenitor alveolar cells and differentiation on lung microscaffolds
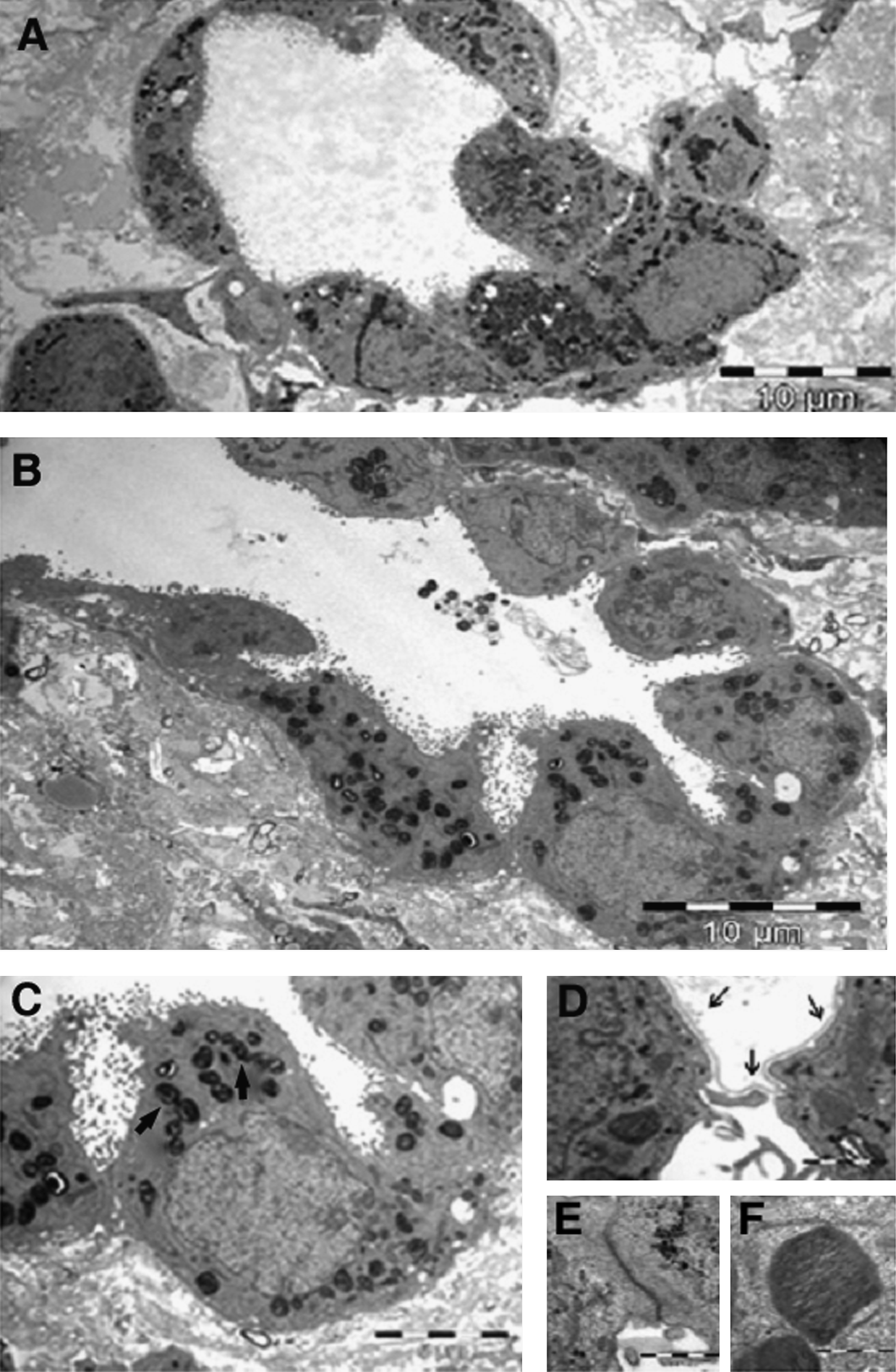
When monolayer-expanded adult AP cells were seeded on the lung microscaffolds, the cells integrated and, with time, continued to proliferate and expand within the scaffolds. Histological sections revealed that the AP cells covered not only the outer surface of the lung
AP cells are known to dedifferentiate with time when grown on standard monolayer cultures (ref. 15 and Fig. 5A–C). In contrast, AP cells seeded on lung-derived microscaffolds were found to transcribe the water channel protein aquaporin 5 (AQP-5) and the transmembrane protein T1α within 7 days (Fig. 5A). These markers are specific for type I cells. CC10 which is a marker for Clara cells was only expressed when AP cells were seeded into lung-derived microscaffolds immediately after preparation, but not after expansion (data not shown). In contrast, the gene coding for surfactant protein C (SPC), which is specific to type II cells, was induced in the adult progenitor cells seeded on lung-derived microscaffolds and its expression level increased with time (Fig. 5A, B). Further, as shown in Figure 5C, AP cells that were grown for two passages in monolayer culture prior to seeding onto lung-derived microscaffolds and cultured for a further 22 days also displayed de novo transcription of SPC. To our knowledge this is the first report describing a system in which AP cells can retranscribe sustained levels of surfactant genes after long periods of monolayer culture, wherein this trait was lost.
Repopulation of lung microscaffolds with adult progenitor alveolar (AP) cells.
Transmission electron microscopy of lung-derived microscaffolds that have been seeded with AP cells and cultured for 14 days.

Lung-derived but not liver-derived microscaffolds induce the expression of alveolar-specific genes in adult progenitor alveolar (AP) cells.
Organ-derived scaffolds show tissue specificity
To clarify the extent of specificity of the scaffold for a particular cell type, we prepared microscaffolds from rat liver organs. AP cells seeded on liver-derived microscaffolds and cultured for 14 days failed to transcribe SPC and AQP-5 (Fig. 5D). Not surprisingly, AP cells did transcribe T1α when seeded onto liver-derived microscaffolds, as this gene was also transcribed by the AP cells in monolayer cultures (Fig. 5A, D). Conversely, primary rat hepatocytes seeded on liver-derived microscaffolds transcribed the liver-specific genes coding for albumin, coagulation factor V (F5), glycogen synthase 2 (GS), glutathione S-transferase α3 (α-GST), and major urinary protein-1 (MUP) when cultured for 14 days (Fig. 5E). Hepatocytes grown on lung-derived microscaffolds were found to transcribe albumin and coagulation factor V, but failed to express GS, α-GST, and MUP (Fig. 5E). It should be noted that hepatocytes in standard monolayer cultures do express albumin 16 and clotting factors. 17
The lung-derived microscaffolds support the spatial pattern of surfactant expression
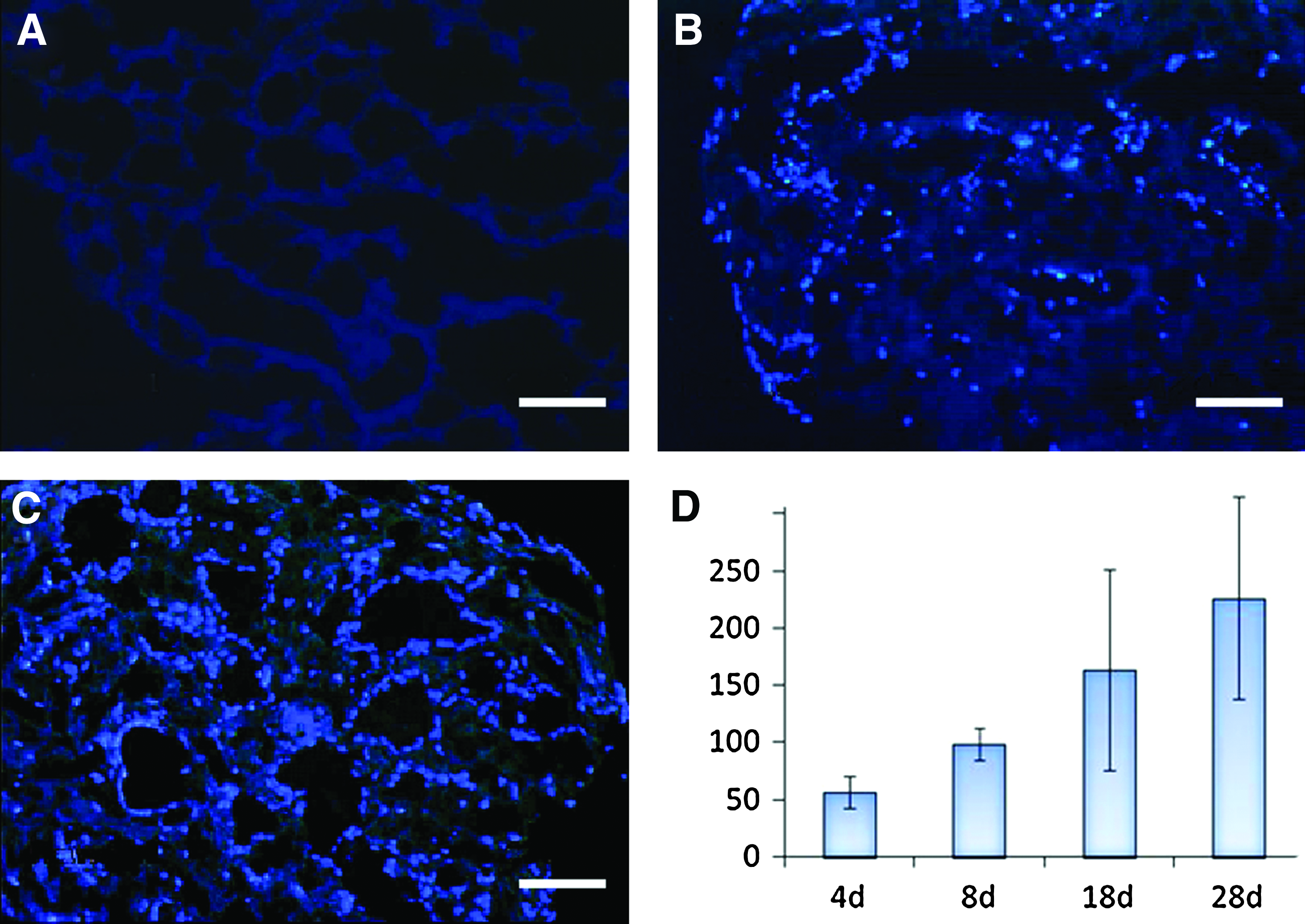
We next determined the spatial pattern of surfactant expression within the newly formed alveolar structures of AP cells in lung microscaffolds. As shown in Figure 6, pro-surfactant protein B (pro-SPB) was found to be restricted to cells lining the alveolar cavities (Fig. 6A, B). About 15%–30% of these cells expressed pro-SPB (Fig. 6A, B). Moreover, the cells that expressed pro-SPB had a rounded morphology typical of type II alveolar epithelial cells (Fig. 6D). Altogether, our study shows that AP cells seeded in lung-derived microscaffolds form highly organized alveolar epithelia containing at least two morphologically and biochemically distinct types of cells that are highly reminiscent of the arrangement of the normal alveolar lung (Fig. 6C).
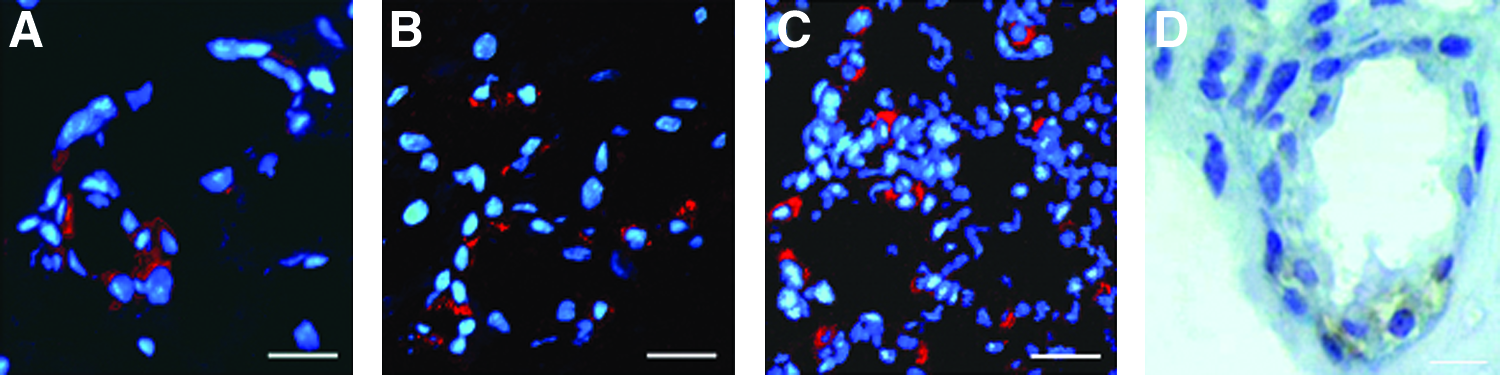
Adult progenitor alveolar (AP) cells differentiate and form highly organized structures in lung microscaffolds. Cryostat, transverse sections of AP cells cultured on lung-derived microscaffolds for 27 days.
Differential composition of lung and liver scaffolds
In an attempt to elucidate the molecular signals involved in this remarkable instructive differentiation process, we have characterized the protein composition of rat lung- and liver-derived microscaffolds by capillary chromatography-tandem mass spectrometry. Proteomic analysis indicates that organ scaffolds are highly complex and the lung microscaffolds contain at least six different collagen chains, six types of laminins, and several growth factors in different proportions (Table 1 and Supplementary Table S2). We found 26 differentially expressed peptides between the two organ sources (Table 1). The most pronounced ones based on the richness and diversity of the laminin genes were six isoforms in lung when compared with only two in liver microscaffolds. Especially laminin alpha proteins are enriched in the lung scaffolds compared with the liver scaffolds (Table 1). Laminin alpha4 and alpha5 detected in our lung scaffolds are in agreement with previous findings.18,19 In contrast to those reports, we did detect laminin alpha2 at medium levels. In both lung and liver tissues, a large fraction of the ECM is based on COL1A1 and COL2A1. Other key players in cell-to-cell and cell-to-matrix interactions, such as thrombospondin 1, vimentin, and decorin, show also differential expression (Table 1). To our knowledge, this is the first attempt of making a comprehensive comparative analysis of extracellular matrix-related proteins presented in lung and liver scaffolds. Still, there is a need for a higher resolution of this analysis, as we are aware that rarer components were only detected at a lower level of certainty. Of these, only the important components of collagen type IV of both lung and liver basement membranes19,20 were included in Table 1 and Supplementary Table S2.
Components in bold are expressed at higher levels in lung than in liver microscaffolds.
Discussion
In this article, we described a novel 3D culture system based on natural organ-specific microenvironment that provides the optimal conditions for expanding primary cells. This system has the advantage of being built up by the 3D organ-specific fine structure and having a microscopic thickness, such that no vascularization is required. These natural microscaffolds not only supported cell growth of primary cells but also their differentiation. The long-term function of progenitor alveolar cells when grown in lung, but not liver microscaffolds, emphasizes the importance of the quality and dimensions of the scaffold chosen. Two different sources of scaffolds were examined to determine the extent of functional specificity of each scaffold for a particular cell type. Although hepatocytes survived well in lung-derived microscaffolds and in liver-derived microscaffolds, all liver-specific genes tested were found to be expressed only in liver-derived microscaffolds (Fig. 3D). In contrast, highly organized alveolar structures were obtained only when AP cells were cultured in lung-derived microscaffolds (Fig. 4), but not when cultured in monolayer cultures or on liver-derived microscaffolds (data not shown). It should be emphasized that the alveolar cell function was retrieved in our microscaffolds even though the progenitor cells were obtained from adults. It is worthwhile to point out that adult-derived scaffolds retain sufficient information to instruct such degree of specificity.
A significant amount of effort has been invested in determining to what extent the stroma affects the fate of epithelial cells during development.21–24 Data presented here suggest that a similar mechanism may take place in the adult. This is in full agreement with the emerging concept that adult tissues in some way recapitulate development.25,26 As shown in Figure 4, AP-derived cells in disparate regions of the same one-layer-thick contiguous epithelium displayed distinct phenotypes. The fact that such a remarkable degree of organization was obtained at the single-cell level strongly suggests that signals within the scaffold are highly localized. In fact, a physical scaffold may well be a reliable vehicle to create or maintain pattern at microlevel resolution.
Recently, several research groups have described a similar decellularization–recellularization procedure to engineer whole-complex organs intended for transplantation.27–32 Petersen et al. 27 and Ott et al. 30 managed to prepare lung scaffolds that retain the hierarchical branching structures of airways and vasculature. When a mixed population of lung epithelial cells and lung microvascular endothelial cells were seeded onto these acellular scaffolds, a hierarchical organization within the matrix was obtained whose mechanical characteristics were similar to those of native lung tissue. When implanted into rats for short time intervals (up to 6 h), the engineered lungs participated in gas exchange.27,30 Whole-engineered organs suffer from the lack of suitable circulation required for gas and nutrient exchange. 33 Attempts have been made to incorporate artificial capillary networks into synthetic matrices. Fidkowski et al. 34 described an approach where capillary patterns are etched by standard microelectromechanical techniques. The resultant silicon wafers are then used as micromolds to prepare a capillary network of polyglycerol sebacate. Another group has recently built a microfluidic chip coated with lung cells and blood capillaries in a predefined geometrical pattern that mimics a human microlung resulting in a biologically relevant model for testing drugs or conducting toxicity screens. 35 Petersen et al. 36 claimed that large segments of lung tissue can only be studied ex vivo for up to a couple of hours in the laboratory, unless using a sophisticated bioreactor that can support lung tissue for up to 1 week. Our natural ECM 3D culture platform overcomes this problem, as it recognizes the importance of small dimensions. In light of the ability of organ-specific microscaffolds to support cell function for extended periods, it can be easily used for studying pulmonary and liver biology in vitro and as a vehicle for in vitro primary cell expansion.
Lung-derived microscaffolds allow for simple in vitro experimentation, so that conditions required for proper differentiation and function of alveolar cells can be monitored. A serious limitation in any attempt to regenerate an artificial lung is the lack of autologous cells for organ reconstitution and eventual transplantation. Thus, there is a need for obtaining large numbers of differentiated alveolar cells. We show here that adult lung cells can be expanded in standard monolayer cultures, which can then be differentiated into type I and type II alveolar cells when seeded on lung-derived microscaffolds. Further, we show that cell differentiation of AP cells in lung microscaffolds into type II cells, as in the normal lung, seems to be highly specific and restricted to distinct areas within each alveolus.
It should be noted that, in our model, AP cells that have been expanded in monolayer culture and ceased to transcribe surfactants redifferentiate when seeded on lung-derived microscaffolds. Because of the tiny dimensions, AP cells may grow in the microscaffolds for weeks. Moreover, the AP cells express a different gene repertoire depending on which microenvironment they encounter (Fig. 3C). Altogether, we have chosen to use a relatively simple system that did not include endothelial cells as used by Petersen et al. 27 and Ott et al. 30 Yet, a significant degree of organization and alveolar cell differentiation was obtained, suggesting that information stored in the lung microscaffolds is sufficient to instruct for proper alveolar localization and differentiation. Despite this simplification, deciphering the molecular nature of the signals involved is complex. Most likely the degree of organization and cell differentiation observed is the result of a combination of signals distributed in a clearly defined spatial pattern within the microscaffold. Based on the large complexity of each different type of stroma, it comes as no surprise that expanded lung cells only differentiate when presented within the appropriate complex microenvironment. By comparing the ECM components of different scaffolds, it may be possible in the future to determine which components are necessary for the precise degree of localized differentiation of the seeded cells reported here.
Conclusions
We have here described a novel 3D in vitro system for the preparation of engineered tissues, based on natural decellularized organ-specific scaffolds of microscopic dimensions, which do not require vascularization. We show that the organ-specific scaffolds instruct the differentiation of primary cells derived from the same organ. Thus, these microscaffolds are an ideal system for allowing expanded cells to display in vivo-like tissue-specific differentiated functions in vitro.
Footnotes
Acknowledgment
This work was supported by a grant from the Alston Fund to E.M.
Disclosure Statement
The broad platform covering the in vitro culture system described here has been patented. A large pharmaceutical company has licensed several applications of the technology. There has been no financial support for the work presented here that could have affected the results.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.