
Abstract
Glycosaminoglycans, like heparin, are frequently incorporated in biomaterials because of their capacity to bind and store growth factors and because of their hydrating properties. Heparin is also often used in biomaterials for its anticoagulant activity. Analysis of biomaterial-bound heparin is challenging because most assays are based on heparin in solution. In this study, seven different methods were probed to analyze heparin covalently attached to collagen scaffolds. For each method, the basic mechanism and the advantages and disadvantages are given. An analysis by the factor Xa assay and the Farndale assay clearly indicated that the amount of immobilized heparin cannot be determined correctly when the scaffold is intact. Scaffolds had to be proteolytically digested or acid treated to obtain reliable measurements. Methods used to quantify the amount of bound heparin included a hexosamine assay, an uronic acid assay, a Farndale assay, agarose gel electrophoresis, and immuno-dot blot analysis. Location and semiquantification of heparin were accomplished by immunofluorescence. Although all assays had their advantages and disadvantages, the hexosamine assay turned out to be the most robust and is recommended as the preferred assay to quantify the amount of heparin bound to scaffolds. It is applicable to all scaffolds that are acid hydrolyzable. This study may allow researchers in the field to select the most appropriate method to analyze glycosaminoglycans in biomaterials.
Introduction
In tissue engineering, heparin is the most employed glycosaminoglycan for a number of reasons. It is well known for its anticoagulant activity and therefore used in vascular grafts to prevent thrombus formation. 4 Heparin is relatively cheap because it is prepared in large quantities. Immobilized heparin can be used to noncovalently bind growth factors, thus avoiding the risk of loss of growth factor activity, which may occur when they are covalently bound to biomaterials. 5
Heparin consists of highly sulfated disaccharide units (Fig. 1). In intestinal mucosa, a common source of commercially available heparin, about 75% of the disaccharides consist of a 2-O-sulfated iduronic acid residue and a 6-O-sulfated, N-sulfated glucosamine residue. 6
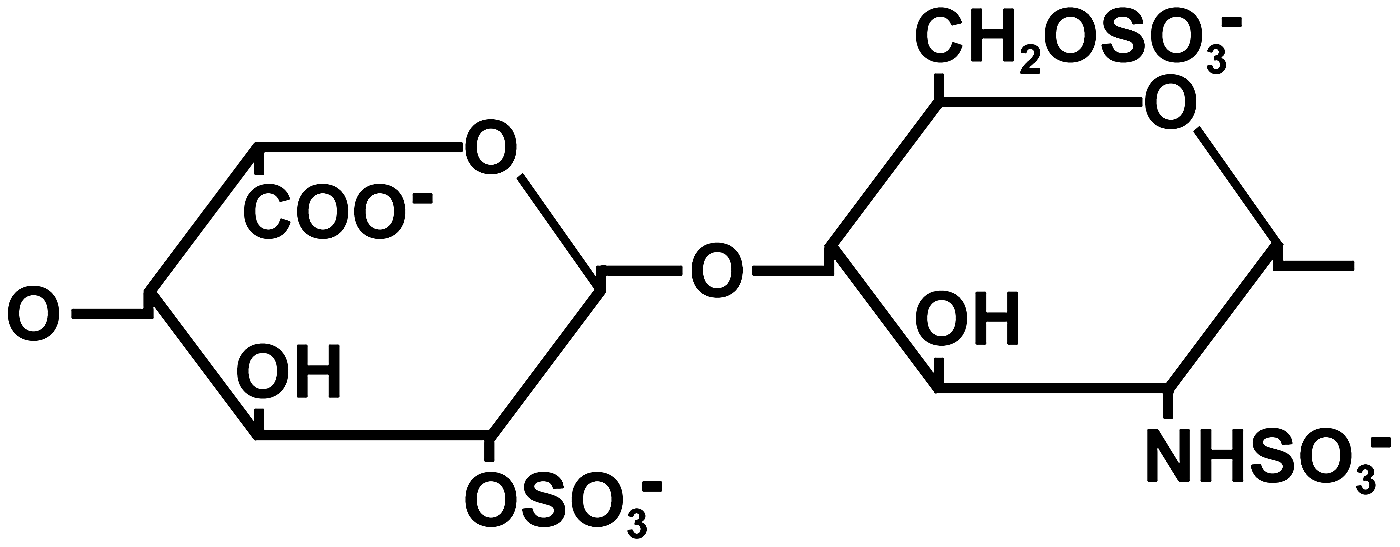
Frequently occurring disaccharide present in heparin from the intestinal mucosa and consisting of a 2-O-sulfated iduronic acid (left) and a 6-O-sulfated, N-sulfated glucosamine residue (right). 6
When immobilizing heparin to a biomaterial it is important to quantify the amount present. The analysis of heparin covalently bound to a solid substrate such as a collagen scaffold may pose a number of challenges because heparin is generally assayed in solution, and the presence of the insoluble biomaterial may interfere.
In this study, seven different methods were used to analyze the amount and location of collagen scaffold-bound heparin. Of each method, the mode of action is briefly reviewed and the advantages and disadvantages are given to enable researchers to select the most appropriate method to analyze heparin in (collagen-based) biomaterials.
Materials and Methods
Materials
Insoluble type I collagen was purified from bovine Achilles tendon using extractions with aqueous acetic acid, NaCl, urea, and acetone as previously described. 7 Heparin sodium from porcine mucosa (187.4 units/mg), with an average molecular weight of 15 kDa (10–20 kDa), was purchased from Diosynth.
Preparation of (heparinized) collagen scaffolds
Scaffolds were prepared in 24-well plates by freezing 800 μL of a 0.8% type I collagen suspension in 0.25 M acetic acid per well at −20°C and by subsequent lyophilization. 8
To covalently couple heparin, scaffolds were wetted in 50 mM 2-morpholinoethane sulphonic acid buffer (MES, pH 5.0) containing 40% ethanol and preincubated for 30 min in 400 μL of a heparin solution (0%–4%) in MES buffer. Scaffolds were chemically crosslinked for 4 h by adding 400 μL 1-ethyl-3-(3-dimethyl aminopropyl) carbodiimide (EDC)/N-hydroxysuccinimide (NHS) in MES buffer, the final concentrations being 33 and 6 mM, respectively, followed by washings with 0.1 M Na2HPO4, 1 M NaCl, 2 M NaCl, and demineralized water. 9 The final contents of heparin in the crosslinking solution were 0%, 0.05%, 0.1%, 0.2%, 0.3%, 0.4%, 0.5%, 0.75%, 1%, and 2%.
To ensure representative sampling, pie part-shaped pieces from the round scaffolds were assayed, unless stated otherwise.
Determination of amine group content
The amine group content of the scaffolds was estimated spectrophotometrically by using 2,4,6-trinitrobenzene sulphonic acid.10,11 To calculate the amount of amine groups per milligram of collagen scaffold, a correction was applied for the amount of bound heparin, using a hexosamine assay (see later).
Papain digestion of scaffolds
To digest collagen scaffolds, 1 mg was incubated in 2.5 U papain (Sigma-Aldrich) in 1 mL digestion buffer (50 mM NaPO4, 2 mM cysteine, 2 mM EDTA, pH 6.5) for 16 h at 65°C.
To evaluate for the presence of large (undigested) protein fragments, 70 μL of 100% trichloroacetic acid (TCA) per 400 μL sample was added, incubated on ice for 30 min, and then centrifuged at 10,000 g for 20 min at 4°C. No pellet was observed, indicating that the papain treatment was sufficient to digest the collagen scaffolds to soluble protein fragments.
Seven methods used for the determination of heparin in scaffolds
Table 1 provides an overview of different aspects of the seven methods used for the determination of heparin in scaffolds.
o/n, overnight.
Factor Xa assay
This method is based on the conformational change of the serine proteinase inhibitor antithrombin III by heparin, resulting in factor Xa inhibition.12,13 The capacity of scaffold-bound heparin to delay the blood coagulation cascade was assessed using a Coatest Heparin (Chromogenix) according to the manufacturer's protocol. 14 Scaffolds (0.5–1.5 mg) were incubated in 50 μL normal human plasma enriched with 2.5 mU antithrombin III in 50 mM Tris (pH 8.4) for 5 min at 37°C to allow antithrombin III to bind to heparin and undergo a conformational change. Heparin standards (0–100 ng) were used as controls. Then, 25 μL factor Xa was added, mixed, and incubated for 30 s at 37°C to allow activated antithrombin III to inactivate factor Xa. A 50 μL aliquot of a 1 mM chromogenic substrate (S-2222 (Bz-IIe-Glu-(g-OR)-Gly-Arg-pNA·HCl)) was added, mixed, and incubated for 10 min at 37°C, allowing the remaining active factor Xa to hydrolyze the substrate, thereby releasing chromophore p-nitroaniline. The reaction was stopped by the addition of 100 μL of 20% acetic acid and absorbance was measured at 405 nm.
Alternatively, factor Xa assay was performed on papain-digested samples (see Papain digestion of scaffolds section) in the presence of 25 μM antipain (Sigma-Aldrich) to prevent papain from digesting antithrombin III and factor Xa.
Farndale assay
This method is based on a shift in the maximum absorbance wavelength of the dye 1,9-dimethylmethylene blue after binding to multiple sulfate groups (metachromasia). 15 Dry scaffold pieces were incubated in Farndale reagent (43 μM dimethylmethylene blue containing 40 mM glycine and 41 mM NaCl adjusted to pH 3.0 with 2 M HCl) and incubated at room temperature.
Alternatively, papain-digested scaffold preparation after TCA precipitation was used (see Papain digestion of scaffolds section). Glycosaminoglycans present in the supernatant (bound to small protein fragments) were analyzed by precipitation with 2 mL ice-cold ethanol, overnight incubation at −20°C, and centrifugation at 17,000 g for 20 min at 4°C. The precipitate was dissolved in 100 μL MilliQ and 10 μL was used for analysis. Heparin standards (0–5 μg) were included and 1 mL Farndale reagent was added to the samples and the standards. The absorbance of dimethylmethylene blue after binding with the sulfate groups in heparin was measured at 525 nm. 16
Hexosamine assay
This method is based on the colorimetric determination of hexosamines, such as glucosamine.17,18 Scaffolds (0.5–4 mg dry weight) and heparin standards (0–500 μg) were hydrolyzed with 500 μL of 6 M HCl at 105°C for 6 h under nitrogen gas in sealed glass tubes. HCl and water were removed by drying the hydrolyzed samples in a vacuum desiccator in the presence of NaOH pellets. Samples were dissolved in 1.25 mL MilliQ, and to 1 mL sample, 1 mL of 4% acetyl acetone in 1.25 M Na2CO3 was added and incubated at 95°C for 1 h to convert the glucosamines to pyrroles. Then, 5 mL of 95% ethanol was added, followed by 1 mL Ehrlich reagent containing 2.66% p-dimethylamino-benzaldehyde in 3 M HCl and 47.5% ethanol to condense pyrroles to a colored product. Absorbance was measured spectrophotometrically at 527 nm.
Uronic acid assay
This method is based on the conversion of uronic acids to a colored product. 19 First, scaffolds (0.5–3 mg) were hydrolyzed in 1 mL of hot 83% H2SO4 containing 120 mM sodium tetraborate in a 48-well plate in a water bath at 80°C for 45 min. Heparin standards (0–150 μg) were used as controls. Uronic acids (both iduronic acid and the less frequently present glucuronic acid) were transformed to a colored product by the addition of 200 μL of 0.2% m-hydroxybiphenyl in 78.4% H2SO4 at room temperature for 15 min and measured at 450 nm.
Agarose gel electrophoresis
This method is based on backbone-dependent separation of glycosaminoglycans in an agarose gel and subsequent staining with the cationic dye azure A and silver as described by Van De Lest et al. 20 Papain-digested scaffold preparation (see Papain digestion of scaffolds section) was diluted 5× in MilliQ water and 1 μL was loaded on 1% agarose gels. To enable quantification, heparin standards of 5, 10, 20, and 40 ng were included. Electrophoresis was performed in 50 mM barium acetate buffer (pH 5.0) at 60 V for 50 min. The gels were subsequently fixed and prestained in 0.1% (w/v) azure A in 50 mM sodium formate and 10 mM magnesium chloride (pH 3.5). The gels were decolorized with 10 mM sodium acetate buffer (pH 5.5, 2×30 min) and demineralized water (30 min). After drying in air, the gels were rehydrated in MilliQ water and silver stained in 15 mM AgNO3, 1.75 mM silicotungstic acid, 75 mM formaldehyde in 30 mM NH4NO3, and 0.24 M Na2CO3. The staining reaction was ceased by incubating the gels in demineralized water for 2 min, in 1% acetic acid for 5 min, and in demineralized water for 15 min. The gels were air-dried and scanned for analysis. For quantification, the median of the pixel intensity of each band was determined with an equally sized selection area using Adobe Photoshop (Adobe Systems). Values were corrected for background intensity using an adjacent area, and staining differences between gels were corrected using a 5 ng heparin standard loaded on each gel.
Immuno-dot blot
This method is based on antibody recognition of heparin, bound to a nitrocellulose membrane by virtue of small collagen fragments still attached to the heparin. Of a papain-digested scaffold preparation (see Papain digestion of scaffolds section), 50 μL was transferred to a Hybond-ECL nitrocellulose membrane (Amersham Biosciences) using an Easy-Titer enzyme-linked immunoflow assay system (ELIFA; Pierce Biotechnology). After 30 min at room temperature, nonspecific binding was blocked with 1% bovine serum albumin (BSA) in phosphate-buffered saline (PBS, pH 7.2) for 16 h at 4°C. Membrane-bound collagen peptide–heparin was detected using the phage display-derived VSV-tagged antibody HS4C3 (1:50), which recognizes heparin, 21 followed by mouse anti-VSV tag antibody P5D4 (1:10), and a peroxidase-conjugated goat anti-mouse IgG antibody (1:20,000; Pierce), all in 1% BSA in PBS, followed by the ECL Western Blotting Detection kit (Amersham Biosciences). Scanned images of exposed films were used for determination of the relative pixel intensity. Pictures were inverted, and the median of the pixel intensity of equally sized areas containing one spot was determined using Adobe Photoshop. Values were corrected for background intensity using a spot adjacent to the spot of interest and expressed as a percentage between background intensity (0%) and complete saturation (100%).
Immunofluorescence assay
This method is based on the antibody recognition of heparin in scaffold sections. Scaffolds were carefully wetted in demineralized water, placed in TissueTek (Sakura Finetek), and snap-frozen in liquid nitrogen–cooled isopentane. Cryosections of 5 μm thickness were cut, placed on SuperFrost slides (Menzel-Gläser), and air-dried for 16 h at room temperature. Nonspecific binding was blocked using 1% BSA in PBS for 10 min and sections were stained with antiheparin antibody HS4C3-VSV (1:5–50), followed by mouse anti-VSV tag antibody P5D4 (1:10), and an Alexa 488-labeled goat anti-mouse IgG antibody (Molecular Probes; 1:100), all in 1% BSA in PBS at room temperature. Staining was evaluated using a Leica DM6000 microscope.
Results
Porous collagen scaffolds of ∼5 mm thickness were prepared in 24-well plates by lyophilization of a frozen collagen suspension. Scaffolds were chemically crosslinked in a solution containing a fixed concentration of EDC/NHS and different amounts of heparin (0%–2%) and then washed extensively.
The amount of immobilized heparin was initially determined by factor Xa assay on intact scaffolds (Fig. 2A) and on papain-digested scaffolds (Fig. 2B). The shape of both curves was comparable and showed an increasing amount of incorporated heparin starting from 0.4% heparin in the crosslinking solution to a maximum at about 1%. However, a 500-fold difference was observed in the absolute values obtained (e.g., 0.022 units in intact scaffolds versus 10 units in digested scaffolds). Apparently, not all immobilized heparin can be measured in intact scaffolds, indicating the need for solubilization of scaffolds for a reliable measurement.

Analysis of immobilized heparin by the factor Xa assay. Anticoagulant activity and deduced amount of immobilized heparin after crosslinking of collagen scaffolds in the presence of 0%–2% heparin in the crosslinking solution, determined for intact
This was also seen when scaffolds were assayed using the Farndale reagent. Incubation of intact scaffolds in the staining solution did result in metachromasia (i.e., a change from a blue to a purple color; Fig. 3), thereby demonstrating the presence of heparin (the scaffolds without heparin are blue), but the intensity of the purple color did not correspond to the amount of heparin immobilized (as assayed by hexosamine assay; see later). Together with the factor Xa data, this clearly indicates that the amount of heparin cannot be assayed correctly when the scaffold is intact. We did, however, notice that the blue staining intensity of the surrounding solution negatively correlated with the amount of heparin immobilized. Therefore, this approach may be suitable for an indirect semiquantitative determination of the amount of scaffold-bound heparin, but the development of standards will be challenging and we did not pursue this strategy.

Analysis of immobilized heparin by the Farndale assay. Scaffolds containing 0, 38, and 167 μg heparin/mg (based on hexosamine assay results; see later) were incubated using the Farndale reagent. Scaffolds containing heparin had a different color than scaffolds without heparin, but no difference was observed between different amounts of bound heparin. Color images available online at
Using papain-digested, solubilized scaffolds, differences in the amount of heparin could be determined with the Farndale assay (Fig. 4A). The shape of the curve, however, differed from the Xa assay, with an increasing amount of incorporated heparin from 0% to 0.4% heparin in the crosslinking solution, to a decrease with increasing heparin content in the crosslinking solution.

Analysis of immobilized heparin assayed with
Heparin was also assessed by measuring specific monosaccharides obtained after acid solubilization of the scaffolds. The shape of the curves obtained by a hexosamine assay and an uronic acid assay were generally comparable to that obtained by the Farndale assay, that is, an initial increase of the amount of heparin immobilized, followed by a decrease (Fig. 4B, C). However, a difference was observed in absolute numbers [e.g., the peak values, ranging from 60 μg/mg (Farndale, A) and 119 μg/mg (hexosamine, B) to 171 μg/mg (uronic acid assay, C; see Discussion section)]. In the uronic acid assay, a heparin-dependent brown color was observed in the samples containing scaffold collagen, but not in the samples containing heparin only (for the standard curve).
The hexosamine assay appeared to be the most robust assay for the quantitative determination of scaffold-bound heparin possibly because, in contrast to the other assays, no delicate precipitation steps were required and no collagen interference with the measurements was observed.
Agarose gel electrophoretic analysis of heparin using papain-digested scaffolds gave variable results in our hands. Staining quality varied and therefore not all gels were suitable for quantification. Results (Fig. 5) indicated a trend comparable to the Farndale, hexosamine, and uronic acid assays, i.e., an initial increase of heparin immobilized followed by a decrease. The peak value observed in the gel presented in Figure 5 (0.3% heparin in crosslinking solution) was about 224 μg heparin/mg. Please note that as one gel contained 7–8 slots, two gels were required to load all standards and samples once, which may limit accuracy.

Analysis of immobilized heparin by agarose gel electrophoresis. Quantification (μg heparin/mg scaffold) based on heparin standards. Please note differences in background staining that may complicate quantification. This figure is a composite picture of two gels (gel 1=0–0.2, gel 2=0.3–2).
Immuno-dot blot analysis of papain-digested samples provides comparable results (Fig. 6). In line with Kreuger et al., no staining was observed when free heparin was applied to the membrane. 22 As a result, no heparin standards could be applied for quantification purposes, but the relative staining intensity peaked at 0.3% heparin in the crosslinking solution.

Analysis of immobilized heparin using an immuno-dot blot assay on papain-digested scaffolds, crosslinked with 0%–2% heparin in the crosslinking solution, and incubation with antiheparin antibody HS4C3. The relative pixel intensity of each spot was measured. No absolute amount of heparin could be calculated, as standards of free heparin do not bind to the membrane.
Finally, an immunofluorescence assay (Fig. 7) was applied to obtain additional information on the location of the immobilized heparin. Heparin was mainly localized at the scaffold surfaces for scaffolds crosslinked in the presence of 0.05% heparin. Scaffolds crosslinked in the presence of 0.4% heparin showed a more even distribution, whereas for scaffolds incubated in 1.0% heparin, less-intense staining was observed (especially at the inner side). These scaffolds contained 38, 80, and 58 μg heparin/mg, respectively (as determined with a hexosamine assay). Therefore, although semiquantitative, results indicate the same trends as observed in Figures 4–6.
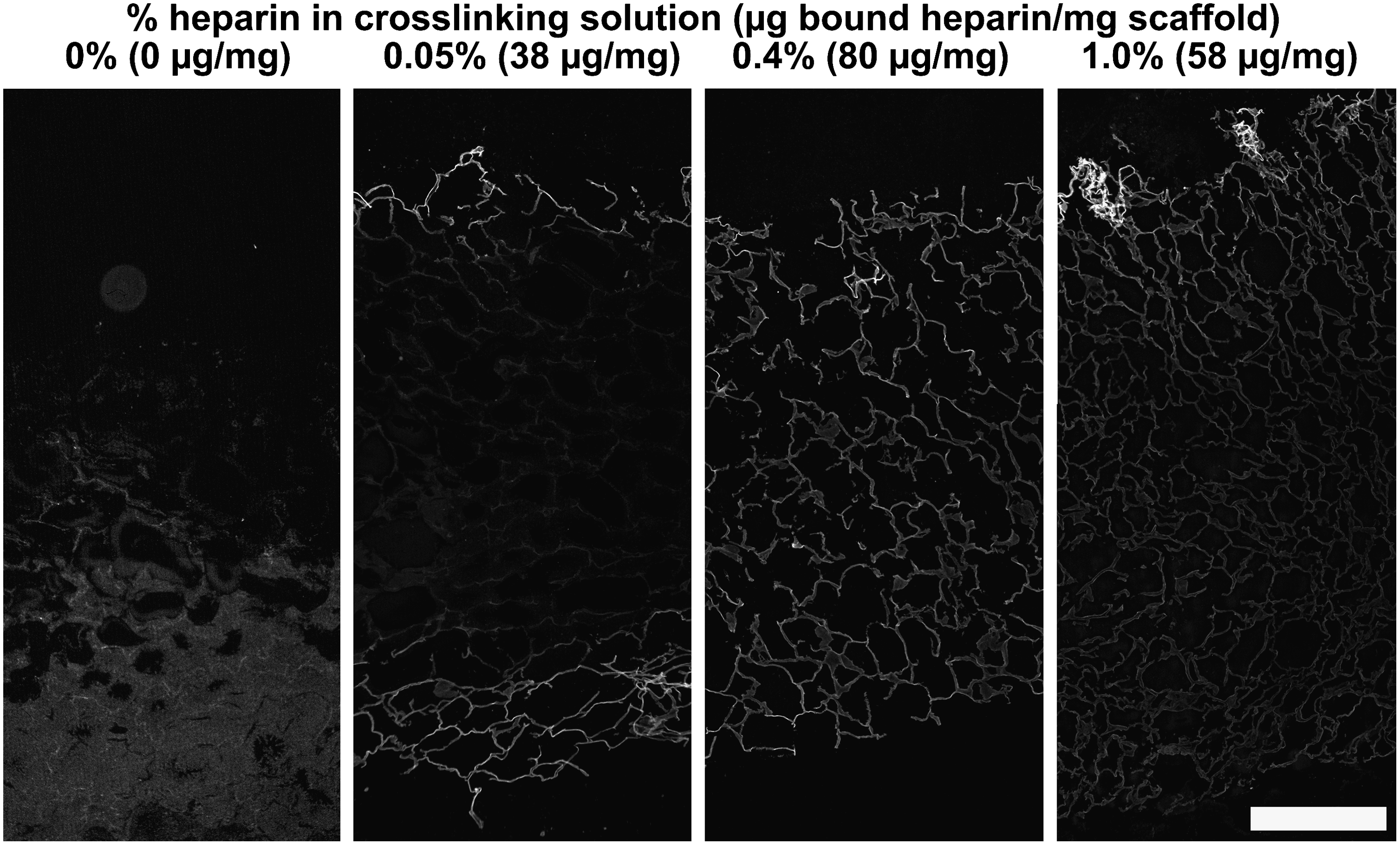
Analysis of immobilized heparin by an immunofluorescence assay. Scaffolds crosslinked in the presence of 0, 0.05, 0.4, and 1.0% heparin were evaluated. The amount of bound heparin as determined with a hexosamine assay on individual scaffolds is given in μg/mg scaffold. Crosslinking in the presence of 0.05% heparin resulted in staining mainly localized at the scaffold surfaces, whereas crosslinking in the presence of 0.4% heparin resulted in a more even distribution of heparin throughout the whole scaffold. Crosslinking in 1.0% heparin showed less-intense staining of primarily the inner area, indicating the same trend as observed in Figures 4–6. Composite pictures were all taken with identical microscope settings. Scale bar is 500 μm.
To find an explanation for the shape of the curves seen in the aforementioned assays (Figs. 2 and 4–7), we evaluated the content of primary amine groups still present after the crosslinking process with respect to the percentage of heparin present in the crosslinking solution. Noncrosslinked scaffolds contained 270 nmol amine groups per milligram, and this was reduced to ∼150 nmol/mg when crosslinking took place in the presence of 0%–0.4% heparin. Crosslinking in the presence of higher amounts of heparin resulted in a higher number of remaining amine groups, even though the EDC concentration was constant for all crosslinking solutions. This may provide clues for the explanation of the results of the other assays (see Discussion section).
Discussion
In this study, seven assays were evaluated for their suitability to quantify the amount of heparin immobilized to insoluble collagenous scaffolds. Both the factor Xa assay and the Farndale assay indicated that quantifying heparin when the scaffolds are intact results in a gross underestimation of the amount of heparin present. For the factor Xa assay, an ∼500-fold difference was noticed, when intact and papain-digested scaffolds were measured, suggesting that in an intact scaffold not all heparin is accessible. Therefore, scaffolds were solubilized either using papain digestion or by acid hydrolysis.
The hexosamine assay turned out to be the most robust assay and is therefore the preferred method for the quantification of immobilized heparin, despite the relatively long assay time. It digests the scaffold by acid hydrolysis, and it has the advantage over the uronic acid assay that it is not associated with an additional brown coloring. This additional staining is not observed when heparin without collagen is assayed (e.g., for the standard curve) and may therefore result in an overestimation of the amount of heparin immobilized to collagen scaffolds (an up to about 50% higher value was found using the uronic acid assay). The Farndale assay, which uses papain digestion to solubilize heparin, gave lower values compared with the assays that use acid hydrolysis. Papain digestion may result in some (solubilized) heparin being still attached to collagen peptides, which may precipitate in the TCA protein precipitation, thereby leading to an underestimation of the amount of heparin (note that the heparin standards are not TCA precipitated). It is also possible that small heparin-bound collagen fragments interfered with the measurements, as protein (albumin) in the Farndale assay is able to reduce the readout to ∼40%. 16 The inclusion of standards during each step of the assay and spiking experiments may elucidate the effect of the different steps.
The immuno-dot blot method, using an antibody against heparin, gave data that can be compared with each other, but does not result in absolute numbers. The reason for this is that heparin alone will not bind to a nitrocellulose membrane, 22 whereas heparin with some residual amino acid residues (as is the case with papain digestion) does. As a consequence, no free heparin standards could be loaded for quantification. Agarose gel electrophoresis, on the other hand, is not dependent on the presence of peptide residues. It provides information on the backbone structure of the glycosaminoglycan under investigation and can be used quantitatively. A limiting factor is the relative small number of samples that can be assessed on a single gel and this makes high-throughput analysis challenging. Finally, the immunofluorescence assay, although at best semiquantitative, provides information on the location of bound heparin. It was observed that at low heparin concentrations, its location was primarily at the scaffold surfaces, emphasizing the importance of correct sampling.
An interesting feature of the present study is the observation that, after a binding maximum, the amount of bound heparin decreased with increasing heparin concentration in the crosslinking solution. Wissink et al. showed that heparin binding correlated with the ratio of EDC molecules per heparin carboxylic group (EDC:Hep-COOH). 23 When the ratio EDC:Hep-COOH drops (as is the case when the concentration of heparin increases in the presence of a fixed EDC concentration), a point may be reached when heparin molecules may not be sufficiently EDC activated to bind to collagen. This may explain the observed decrease with increasing heparin concentrations and is in line with the observed decrease in the amount of amine groups used for crosslinking (and hence the increase in primary amine groups; Fig. 8). The results of the factor Xa assay, assessing functional anticoagulation, differed from the other assays. In this assay, the accessibility of heparin for the protein antithrombin III (and possibly factor Xa) is of crucial importance, and the way and extent by which heparin is bound to collagen are likely important factors. It has been suggested that an increased number of covalent bonds between heparin and collagen may result in a decreased accessibility of heparin for the antithrombin III binding site and/or decreased thrombin binding. 23 The conditions in our study at low EDC:Hep-COOH ratios (obtained at 1%–2% heparin in the crosslinking solution) may have resulted in less binding of absolute numbers of heparin molecules, but an optimal accessibility of those heparin molecules that did bind, because of the limited number of covalent bonds between collagen and heparin. Maximal exposure of the heparin molecules to antithrombin III may have increased the functional anticoagulation.

Amine group content of the collagen scaffolds after crosslinking in the presence of 0%–2% heparin. The amine group content of noncrosslinked scaffolds was 270 nmol/mg. Results of a typical experiment in triplicate±standard deviation.
The precise conditions used during the crosslinking are likely critical for the amount and mode of binding. For instance, Pieper et al. crosslinked in the presence of 2.75% heparin and reported 129 μg bound heparin/mg scaffold using a hexosamine assay, much higher than expected from our study. 9 They also crosslinked with 33 mM EDC, but used 4 mg scaffold collagen per milliliter of crosslinking solution instead of 8 mg, 20 mM NHS (which forms a stable intermediate during crosslinking) instead of 6 mM, and crosslinked at a pH of 5.5 instead of 5.0, which may explain the observed differences.
Heparin shares many characteristics with other glycosaminoglycans and the methods described here are therefore broadly applicable. For immuno-dot blot and immunofluorescence analysis, additional antibodies that recognize specific sugar modifications present in different glycosaminoglycans are available. 24 All methods may be applicable to a broad range of biomaterials that can be digested by proteases (such as fibrin), hydrolyzed (such as polylactic acid and polycaprolactone), and/or freeze-sectioned.
Conclusion
From seven methods evaluated, the hexosamine assay is the preferred method for the determination of the absolute amount of heparin bound to a scaffold. This robust method may also be applied for other acid-hydrolyzable biomaterials.
Footnotes
Acknowledgments
This study was funded by the Dutch Program for Tissue Engineering (grant DPTE 6735) and by the EU-FP6 project EuroSTEC (soft tissue engineering for congenital birth defects in children: contract: LSHB-CT-2006-037409).
Disclosure Statement
No competing financial interests exist.