
Abstract
Skin has a remarkable capacity for regeneration, but age- and diabetes-related vascular problems lead to chronic non-healing wounds for many thousands of U.K. patients. There is a need for new therapeutic approaches to treat these resistant wounds. Donor mesenchymal stem/stromal cells (MSCs) have been shown to assist cutaneous wound healing by accelerating re-epithelialization. The aim of this work was to devise a low risk and convenient delivery method for transferring these cells to wound beds. Plasma polymerization was used to functionalize the surface of medical-grade silicone with acrylic acid. Cells attached well to these carriers, and culture for up to 3 days on the carriers did not significantly affect their phenotype or ability to support vascular tubule formation. These carriers were then used to transfer MSCs onto human dermis. Cell transfer was confirmed using an MTT assay to assess viable cell numbers and enhanced green fluorescent protein–labeled MSCs to demonstrate that the cells post-transfer attached to the dermis. We conclude that this synthetic carrier membrane is a promising approach for delivery of therapeutic MSCs and opens the way for future studies to evaluate its impact on repairing difficult skin wounds.
Introduction
Bone marrow-MSCs (BM-MSCs) were first discovered in the 1970s, 4 and they have since been shown to differentiate into a wide variety of cell types from different tissues and contribute to tissue repair 5 ; they also provide stromal niche support for both hematopoietic development and neovascularization.6,7 Recently MSCs have been shown to support cutaneous wound regeneration by accelerating wound closure, reducing scarring, and restoring the final tensile-strength of the tissue to levels higher than untreated wounds.8–14 Also their well-documented immunomodulatory and anti-inflammatory properties 15 make these cells an attractive cell source for treating chronic wounds.
The aim of this study was to develop and evaluate a cell carrier to facilitate delivery of MSCs to wound beds. Over the past decade, our group has utilized the technique of plasma polymerization to deposit thin films of acrylic acid on a variety of substrates. We have used these surface modifications to make materials supportive of the attachment and growth of a number of cell types (e.g., keratinocytes, melanocytes, and corneal epithelial cells),16–21 and demonstrated delivery of human keratinocytes 19 and cocultures of keratinocytes and melanocytes17,18 to in vitro model wound beds. These cell carriers have been used successfully in the clinic to deliver autologous keratinocytes for the treatment of patients with extensive skin loss resulting from burn injury and diabetic foot ulcers.20,22–24 Here, we demonstrate for the first time that these carriers are supportive of MSC cultures. The cultures retain their phenotype and ability to support vascular tubule formation after 72 h culture on the carriers, and the carriers allow the effective transfer of MSCs to human dermis model wound beds.
Materials and Methods
Production of plasma-polymerized surfaces (carriers)
Plasma polymerization was carried out using apparatus detailed previously. 25 The reactor was evacuated using a rotary pump, and liquid nitrogen “cold-trap,” to a base pressure of approximately 3×10−3 mbar. Acrylic acid monomer vapor (Sigma-Aldrich, St. Louis, MO) was entered into the system through a needle valve. A flow rate of 4.8–5.2 cm3 (STP) min−1 (sccm) was established and maintained using methods described by Yasuda. 26 A plasma was initiated and sustained using a 13.56 MHz radio frequency power generator inductively coupled to the reactor, with the electrical power being maintained at 2 W. Reflected power was minimized (typically to <0.1 W) by an impedance matching unit. Sheets of medical-grade silicone (Polymer Systems Technology, High Wycombe, England), seated in Petri dishes, were placed in the middle of the reactor for a deposition time of 20 min, before the monomer was allowed to continue to flow through the reactor for a further 10 min. Samples were stored at room temperature and wrapped in Parafilm. All carrier surfaces were used within 60 days.
Carrier surface analysis
X-ray photoelectron spectroscopy (XPS) was carried out using a Kratos Axis Ultra X-ray photoelectron spectrometer (Kratos Analytical, Manchester, England). All samples were maintained under vacuum (1×10−8 mbar) for 2 h prior to analysis, and all data were collected 90° to the sample plane. A minimum of two survey scans and two narrow scans were performed on each sample, with pass energies of 160 eV and 20 eV, respectively, to obtain O1s and C1s peaks. Charge neutralization was applied to all samples. CasaXPS software (
Preparation of deepithelialized dermal substrates
Deepithelialized dermal (DED) substrate was prepared from two sources. Skin was obtained from patients undergoing breast reductions or abdominoplasties with informed consent of the patients and full ethical approval through the Sheffield Hospitals Ethical Committee. All research using this skin took place in the Kroto Research Institute under a Research Tissue Bank Licence No. 12179. When this was not available, Euroskin (glycerol-preserved split thickness skin) was purchased from Euro Tissue Bank, Beverwijk, The Netherlands. Prior to use, the glycerol was thoroughly removed with phosphate buffered saline (PBS). Both fresh skin and glycerol-preserved skin were then treated similarly and incubated in sterile 1 M NaCl for 18 h at 37°C. Subsequently, the epidermis was separated from the dermis to leave behind the DED. These were then incubated at 37°C and 5% CO2 in Green's medium 18 for 48 h as a means to test sterility of the preparation; the medium containing phenol red would rapidly change color, from red to orange, in the presence of any contamination. At the end of this protocol, histology confirmed the largely acellular dermis and that the epidermis was completely removed. The two skin sources were used interchangeably between experiments, and the source is stated in the figure legends.
Mesenchymal stem/stromal cells
Human bone marrow (hBM) MSCs (termed passage 2) were purchased from Lonza Biologics, Slough, England, and expanded in complete mesenchymal stem cell growth medium supplemented with L-glutamine, gentamicin sulfate, amphotericin B, and mesenchymal cell growth supplement (Lonza Biologics). The MSCs were phenotyped initially at passage 4 by fluoroscene activated cell sorting (FACS) for biomarkers as described by Martin-Rendon et al. 28 and as described in “Supplementary Methods and Results” and Appendix Figure 1. In subsequent analyses, MSCs were used between passages 5 and 8. After the cells had reached 80–90% confluence in a T-75 flask, they were detached with 0.25% (v/v) trypsin and 0.02% (w/v) ethylenediaminetetraacetic acid solution in PBS (pH 7.4) (PAA Laboratories GmbH, Pasching, Austria), and seeded in fresh medium onto the carriers or tissue culture plastic (TCP) for further viability, FACS, vascular tubule analysis, and DED transfer studies. For biomarker and viability analysis by FACS, MSCs were seeded at 1×105 cells/well into six-well TCP plates (Costar; Corning Incorporated, Corning, NY) that either contained or did not contain plasma-polymerized acrylic acid (ppAAc) carriers, and cultured for 72 h before detachment with Accutase (PAA Laboratories GmbH). ppAAc carriers were transferred to a fresh six-well plate before cell detachment, to ensure that any cells that may have adhered to the TCP in the well underneath the carriers were excluded from the analysis. Cells were stained for nonviable cells with Topro-3 (Invitrogen, Paisley, Scotland) together with CD90-FITC, CD73-phycoerythrobilin (PE), CD14-PE-Cy7, CD45-peridinin chlorophyll protein (PerCP) (BD Biosciences, Oxford, England), CD105-FITC (R&D Systems, Aylesbury, England), CD34-allophycocyanin (APC) (Miltenyi Biotec, Bagisch-Gladbach, Germany), anti-HLA-ABC-APC, and anti-human leukocyte antigen (HLA)-DR-PE antibodies (Biolegend, Cambridge, England) with appropriate isotype controls. The MSCs were also transduced with an enhanced green fluorescent protein (eGFP) using a lentiviral vector-GFP at multiplicity of infection between 15 and 30 essentially as described previously. 29
Detection of viable MSCs on the carriers
MSCs were seeded at different densities (1×104, 2×104, 4×104, 8×104, and 16×104) onto acrylic acid–coated carriers inside stainless steel rings. Carriers containing cells were incubated for 24 h before the rings and the media were removed. One milliliter of 10% (w/v) Alamar Blue in culture medium (Sigma-Aldrich) was added and the plates were incubated for 3.5 h (37°C, 5% CO2). Subsequently, the Alamar Blue was removed from the culture, and its fluorescence was read (λex=543 nm, λem=590 nm) (Bio-Tek FLx800; BioTek, Potton, England). The culture plates were gently washed with PBS before the fresh culture medium was added. The Alamar Blue assay was also repeated after 7 days of culture.
Vascular tubule assays
Umbilical cord blood was sourced from the John Radcliffe Hospital, Oxford, England, with approval of the NHSBT R&D committee, with ethical approval from the Oxford and Berkshire Ethics Committees, and with fully informed and written consent. Endothelial colony forming cells (ECFCs) were derived from the blood and cultured in complete endothelial growth medium-2 (EGM-2) (Lonza Biologics) as described by Zhang et al., 29 before being phenotyped or labeled with eGFP as described by Khoo et al. 30 About 1×105 hBM-MSCs were seeded into six-well TCP plates that either contained or did not contain ppAAc carriers. Cells were cultured for 24 or 72 h before carrying out vascular tubule assays. ppAAc carriers were transferred to a fresh six well for reasons stated in the “Mesenchymal stem/stromal cells” section. The hBM-MSCs were subsequently reseeded (4×104/well) into collagen-coated 48-well plates (BD Biosciences), and allowed to adhere for 2 h 29 before 2–8×103 (passage 4–5) eGFP-labeled umbilical cord blood ECFC-derived cells were added to the culture in EGM-2. Medium was changed every 2–3 days until day 14 and the wells were photographed. Image analysis was carried out using the semiautomated AngioSys software program (TCS Cellworks, Botolph Claydon, England), which measured the number of junctions and tubules, and total tubule lengths per field (the field area was 8.4 mm2). 30
Cell transfer analysis
About 5×104 cells were seeded onto either untreated silicone or those coated with acrylic acid with an area 2 cm2, using 10-mm-diameter stainless steel rings to confine the cells to the surfaces providing a cell density of 38,000 cells/cm2. Carriers plus cells were incubated (37°C, 5% CO2) for 24 or 72 h, and then transferred to the DED and incubated for a further 24 h. This involved placing the carriers' cell side down on the DED, and placing small stainless steel grids on top of the carriers to hold them in place. The DED plus the carrier was then flooded with media. After 24 h, the carriers and dermis were separated, and the viable cells transferred from the carriers to the DED were assessed by 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT)-eluted stain assay (MTT-ESTA; Sigma-Aldrich) after 24 h, 72 h, and 7 days following cell transfer. The formazan salt formed after 40-min incubation was eluted from the cells with 2-hydroxy ethanol (Sigma-Aldrich, United Kingdom), and the optical densities of the samples were read at 540 nm with a reference filter of 630 nm (Bio-Tek ELx800). The above method was also applied to the eGFP-labeled cells. The MTT assay was performed on blank DED (DED containing no transferred cells); the average optical density (OD) values served as a baseline and were subtracted from the experimental values. In this way, any contribution to the reduction of formazan from the DED alone was accounted for.
Confocal microscopy
All images of eGFP-labeled cells on DED were collected with a Zeiss LSM510 confocal microscope using either a 10× or 40× objective lens (Carl Zeiss, Welwyn Garden City, England). The eGFP was excited with a 488 nm argon laser, and fluorescent light was collected through a 505-nm-long pass emission filter. Z-series compositions were acquired by photographing sections at 2 μm intervals. All cells were imaged at room temperature in prewarmed (37°C) phenol red free alpha minimum essential medium (MEM).
Histology of DED
Samples were fixed in 10% (w/v) formalin in PBS for >24 h and then processed, paraffin embedded, and sectioned to 6 μm thick on a microtome. Sections were stained with haematoxylin and eosin using standard techniques. Collagen IV primary antibody (Abcam PLC, Cambridge, United Kingdom), diluted 1:250 in PBS, was incubated on samples at room temperature for 1 h after heat-mediated antigen retrieval and blocking with 10% goat serum (Sigma-Aldrich, United Kingdom) was performed. For detection, biotinylated goat anti-rabbit secondary antibody, diluted 1:1000 in PBS, was incubated with the samples for 30 min at room temperature and DAB staining was performed using a VECTASTAIN ABC (Vector Laboratories, Peterborough, United Kingdom), according to the manufacturer's instructions.
Statistical analysis
Statistical differences were determined using a two-tailed, unpaired Student's t-test where two groups were compared; one-way analysis of variance, in conjunction with Tukey's test post hoc, was performed where three data sets were compared. Test p-values < 0.05 were regarded as statistically significant.
Results
Characterization of the cell carrier surface
Cell carriers derived from silicone elastomers were prepared by depositing acrylic acid from a low-power (2 W) RF plasma to form a thin surface layer of ppAAc. The XPS analysis was used to analyze the surface elemental and functional composition of the carriers, and surface static WCA were recorded for further characterization. Wide scans revealed the presence of carbon, oxygen, and a small proportion of silicone, indicating that the silicone substrate was covered with a thickness less than the depth of X-ray penetration (8 nm for Si 2p photoelectrons) (Fig. 1A). Narrow scans (high-resolution scans) of the C1s region showed C(=O)OX's high binding energy peak and its associated β-shifted carbon C-C(=O)OX. This is evidence that the surfaces contained carboxyl groups (where X=H) and/or esters (where X=R) and/or anhydride [where R=(O-C(=O)] functionality. The C(=O)OX and C-C(=O)OX functional groups comprised 8.1% and 35.4% of the C1s peak area, respectively (Fig. 1B). The ppAAc coatings also made the surfaces more wettable, reducing the WCA from above 100 to 25.03±3.00, (n=3).
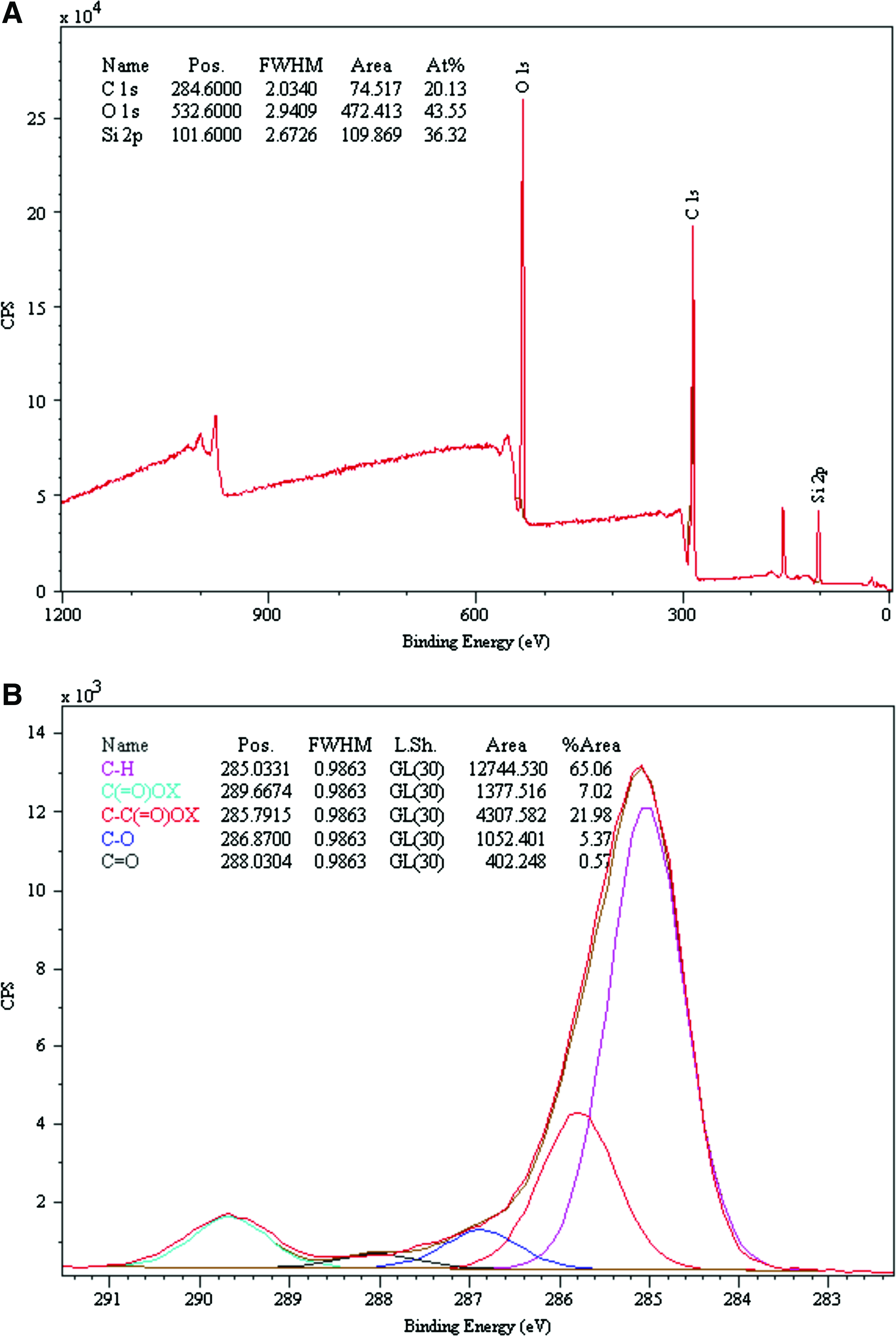
X-ray photoelectron spectroscopy (XPS) of functionalized ppAAc surfaces.
hBM-MSCs remain viable and proliferate on the cell carriers
Flow cytometry confirmed that the hBM-MSCs possessed an antigen profile characteristic of MSCs. Supplementary Figure 1 shows the cells to be negative for CD45, CD14, and CD34, and positive for CD90, CD73, and CD105 (>99% positive), the minimum requirements as specified by the International Society of Cellular Therapy. 31 The cells when supplied had been tested by the manufacturer and by one of our laboratories at passages 4–5 for their trilineage potential 28 (to generate adipogenic, chondrogenic, and osteogenic lineages), and subsequently in our analyses, they were also plastic adherent and had the capacity to support neovascularization in vitro (see Fig. 4). Taken together, the cells had the properties of hBM-MSCs.
Cell viability on the carriers was assessed by Alamar Blue staining 24 h and 7 days after seeding. Several seeding densities were compared to determine the most appropriate cell number for the subsequent cell transfer experiments. The results show that regardless of the seeding density used (1×104 to 16×104), the hBM-MSCs attached to the ppAAc surfaces (carriers) and proliferated over the course of 7 days, in a similar fashion to those cultured on TCP (Fig. 2). However, successive doublings in seeded cell density above 4×104 did not increase cell viability significantly above that of 4×104 cells seeded when determined at day 7. This is likely due to contact inhibition. 32 Accordingly for the subsequent transfer experiments at day 7, cells were seeded at a density of 5×104 per carrier.
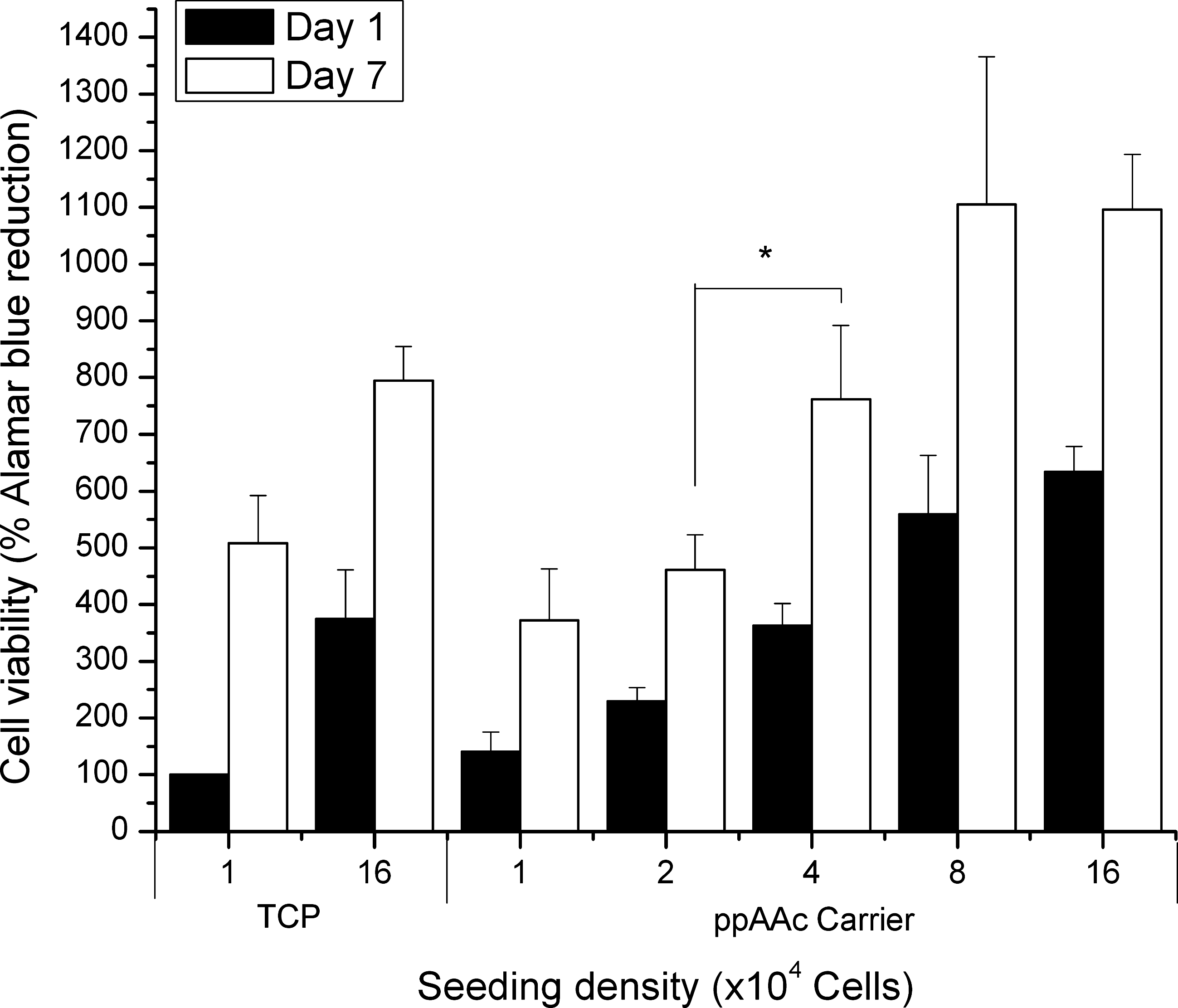
Viable hBM-MSCs on the carrier over the course of 7 days as assessed by Alamar Blue. Cells were seeded at a variety of densities on the carrier ranging from 1×104 to 16×104 cells. The percentage reduction of Alamar Blue is shown normalized to 1×104 cells on tissue culture plastic (TCP). Error bars indicate the SEM for three independent experiments performed in triplicate (*p<0.05). hBM-MSC, human bone marrow mesenchymal stem/stromal cells.
hBM-MSCs maintain their phenotype and ability to support vascular tubule formation after growth on carriers
The hBM-MSCs cultured for 72 h on the carriers or on TCP showed no significant changes in cell viability as measured with Topro-3 staining (Fig. 3A), nor in their cell surface phenotype for key markers CD90, CD105, CD73, CD34, CD14, and CD45 nor for HLA-ABC and HLA-DR expression (Fig. 3B). The hBM-MSCs were also cultured on the carriers and on TCP for 24 and 72 h, and their ability to support vascular tubule formation in coculture with umbilical cord blood ECFC-derived cells in vitro was assessed and quantified. Umbilical cord blood ECFCs do not form tubules when cultured in the absence of hBM-MSCs (Fig. 4A). Cocultures of umbilical cord-blood ECFCs and hBM-MSCs were photographed at day 14 and representative images are shown in Figure 4B. Our results demonstrate that there was no significant difference in the number of junctions formed, number of tubules formed, or total tubule length between hBM-MSCs initially cultured on TCP versus cells initially cultured on the ppAAc carriers for 24 h (p=0.77, p=0.90, p=0.80, respectively) or for 72 h (p=0.37, p=0.88, p=0.69, respectively) (Fig. 4C). There was also no significant difference in the number of junctions formed, number of tubules formed, or total tubule length for cells cultured for 24 h versus 72 h on TCP (p=0.96, p=0.99, p=0.55) or on the ppAAc carriers (p=0.14, p=0.72, p=0.14). Therefore, culture on ppAAc carriers for up to 72 h does not affect the ability of hBM-MSC to support vascular tubule network formation.
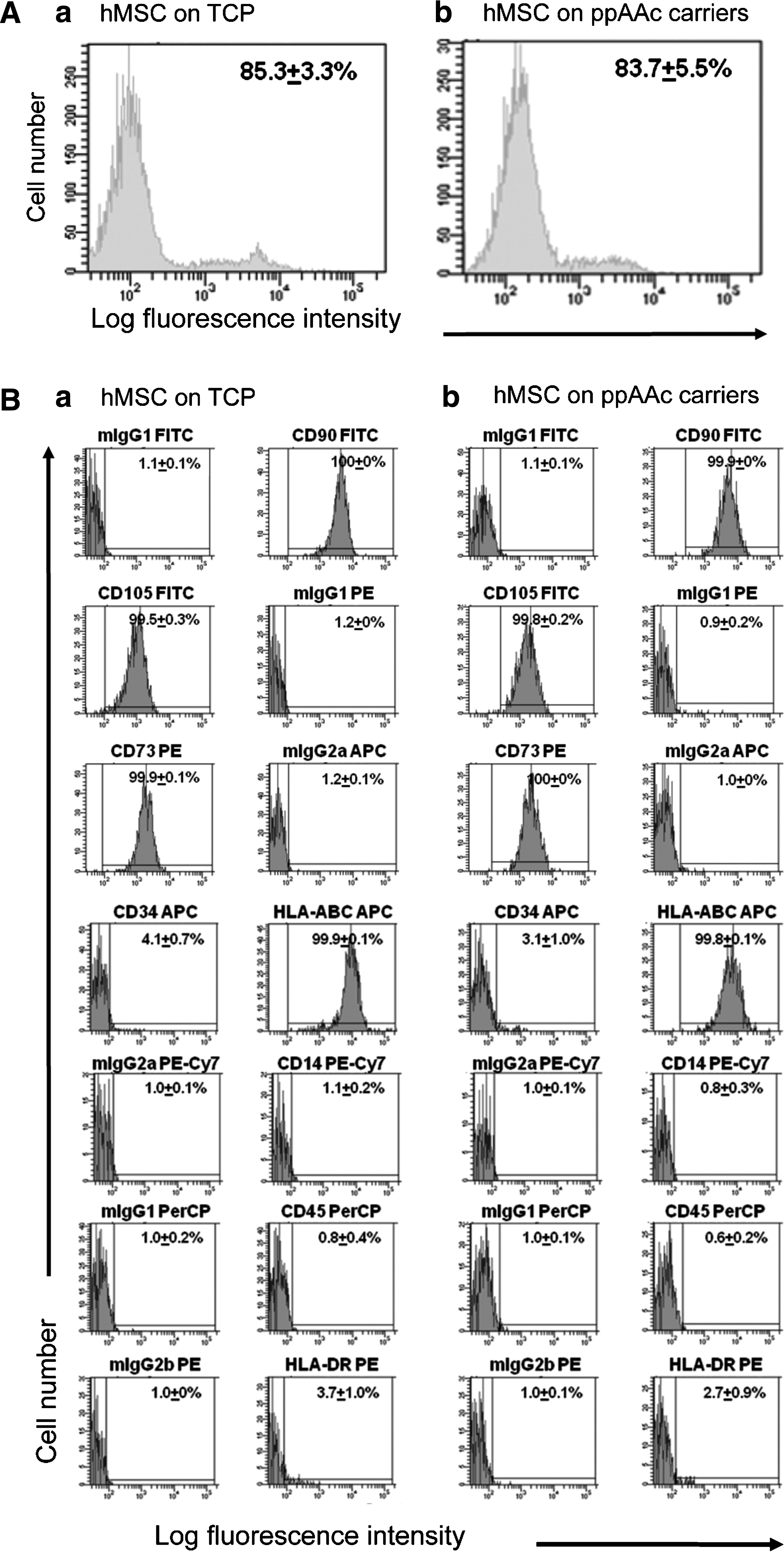
Human bone marrow mesenchymal stem/stromal cells (hBM-MSCs) cultured on ppAAc carriers retain their viability and cell surface phenotype. hBM-MSCs were seeded in TCP plates that did not contain
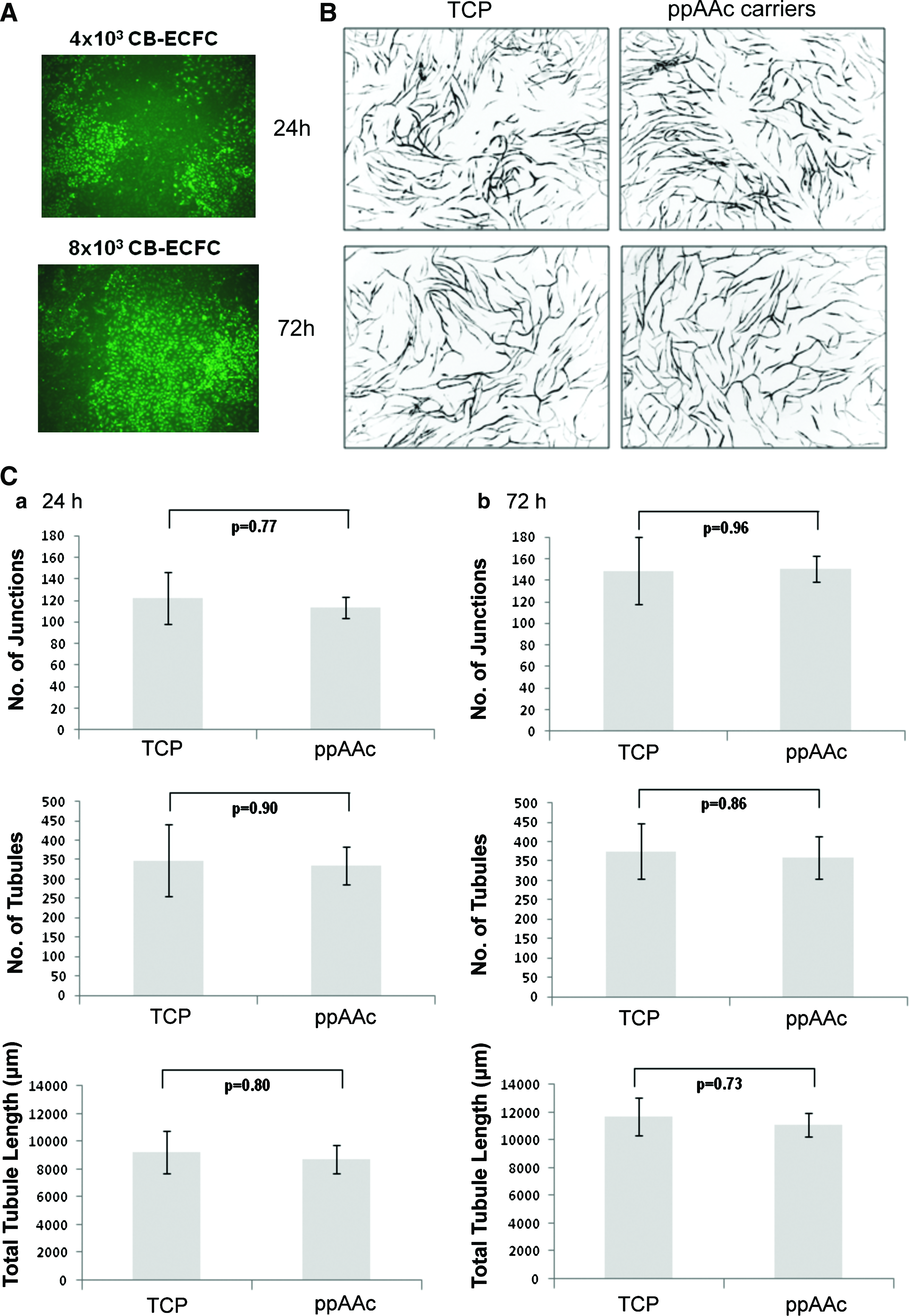
Human bone marrow mesenchymal stem/stromal cells (hBM-MSCs) cultured on ppAAc carriers retain their ability to support vascular tubule formation.
hBM-MSC transfer from carriers to DED
Cells were cultured for 24 h on either unmodified silicone substrates or ppAAc surfaces, before substrates with adherent cells were brought into contact with DED. After the substrates had remained in contact with the DED for 24 h, the carriers were separated from the DED and cell transfer was assessed by MTT assay. The results demonstrate that the cells readily attached to the ppAAc-coated surfaces, whereas cell attachment to the unfunctionalized silicone was very poor (Fig. 5A, B). Consequently, fewer cells were able to transfer from the unfunctionalized surfaces to the surface of the DED as compared with the ppAAc carriers, whereby delivery of a large quantity of cells to the DED was achieved (Fig. 5C, D).

Assessment of cell transfer (ppAAc-coated vs. uncoated carriers) and cell viability post-transfer to deepithelialized dermis by MTT staining.
Subsequently, the effect of the duration of culture on the carriers prior to their transfer to DED were assessed (Fig. 5F, G). First, the hBM-MSCs were cultured on the ppAAc carriers for 24 h and transferred to DED as before, and then cell viability on the DED was measured by MTT assay 24 h, 72 h, and 7 days after transfer (Fig. 5F). Carriers after DED contact were also incubated with MTT to determine whether there were cells remaining on the carrier. This transfer regime was repeated, but it was modified by culturing the cells on the carrier for 72 h before transfer, to determine whether longer periods of culture on the carrier affected cell transfer (Fig. 5G). Figure 5 shows that the cells remained viable post-transfer to the DED for at least 7 days, regardless of the duration of culture on the carrier prior to transfer. Cells from each transfer regime (24 and 72 h on the carrier) also showed a significant increase in viable cells by day 7 (p<0.05), suggesting that the cells had begun to proliferate on the DED after transfer.
There was a slight difference, however, in the degree of cell transfer to the DED after an extended period of culture on the carrier prior to transfer. After 24 h of cell culture on the carrier, few cells remained after transfer (OD values from the carrier were just above the threshold of detection by the plate reader). In contrast, following 72 h of preculture on the carriers, more cells remained on the carrier after transfer to the DED (0.006 vs. 0.012, p<0.05). This was perhaps due to extracellular matrix production by hBM-MSCs enhancing adhesion. Despite this, approximately 80–90% of the cells still transferred to the DED.
While there is a significant difference (p<0.05) between the MTT values obtained for cell viability on the DED between the experiments performed in Figure 5E and F, this may be due to a difference in the metabolic activity of cells from different donors. The key observation is that at all time points cell transfer was good with very few cells remaining on the carriers.
Visualizing cells postdelivery using eGFP-labeled hBM-MSCs
The eGFP-labeled hBM-MSCs were used to trace the cells after transfer. Figure 6A shows that like the nontransfected hBM-MSCs, the eGFP-labeled cells transferred to and proliferated on the DED for at least 7 days after transfer. Confocal microscopy showed that the fluorescent eGFP-labeled hBM-MSCs could be clearly identified before and after transfer (Fig. 6B, C). Interestingly, the morphology of the hBM-MSCs on the carrier and the DED differed. Cells cultured on the carrier had a more “jagged” appearance, whereas those on the DED adopted a more spindle-like morphology. Z-stack images of the DED acquired 7 days after transfer showed cells to be present to a depth of 80 μm (Fig. 6D1, 2, 3, and 4). In addition, histology confirmed the presence of cells on the surface of the DED (Fig. 6E–G). Figure 6E shows the cellular composition of the skin, prior to our NaCl decellularization protocol, consisting of a dense layer of keratinocytes in the epidermis with fibroblasts peppering the dermis (as indicated by the arrows). Following decellularization, these cells cannot be seen (Fig. 6F), and after transfer of cells from the carrier to the DED, a layer of cells can clearly be identified on top of the dermis (Fig. 6G). Figure 6H shows that the basement membrane is still present after decellularization (stained with collagen IV antibody); this could be a barrier to the cells' migration into the DED and may explain why the cells could only be seen superficially on the DED (see Discussion).

eGFP-labeled cells following transfer to deepithelialized dermis.
Discussion
The aim of this work was to evaluate the use of a synthetic cell carrier dressing, prepared from ppAAc-coated medical-grade silicone, for the delivery of MSCs to a skin wound model. Such dressings are currently being used in the clinic to deliver cultured autologous keratinocytes to patients with extensive burn injuries 23 and chronic wounds.20,24 Given the potential therapeutic benefits that MSCs possess with respect to wound healing, it was of interest to determine whether similar surface preparations translate to the efficient delivery of these cells without altering the properties of the cells.
While it is possible to transfer cells using carriers derived from natural materials such as collagen, it is desirable to reduce the use of xenobiotic materials wherever possible, hence our previous work in developing a synthetic carrier. Silicone elastomers are widely used in biomedical applications, such as contact lenses, catheters, heart valves, artificial limbs, and breast implants, because they are considered biocompatable 33 ; however, they are highly hydrophobic and cell attachment is usually (but not always) 34 poor. There are many surface modification strategies that can improve cell attachment to silicone as reviewed by Desmet et al., 35 but they are not without their caveats. Plasma polymerization is a well-established methodology that can provide a defined surface chemistry to almost any substrate with little or no pretreatment, and without detriment to the bulk properties of the material. 35
In this study, we used plasma polymerization, under low-power conditions, to deposit acrylic acid onto medical-grade silicone elastomers with good functional retention. Alexander et al. 27 demonstrated that deposits formed under an energy-deficient regime have an elemental and functional composition similar to that of conventional acrylic acid. The presence of surface acid groups increases the number of oxygen-containing species on the surface, which correlates positively with surface wettability, and in most cases cell adhesion.36,37 Our study showed, as expected, that cell attachment to uncoated silicone substrates was extremely poor, whereas attachment to the more wettable ppAAc-coated substrate was very good (Fig. 5A, B). This result was reflected in the cell transfer data, where far more cells could be delivered to the surface of the DED from ppAAc functionalized surfaces than from nonfunctionalized surfaces (Fig. 5C–E). With regard to the mechanism of transfer, one could speculate that the cells are able to recognize proteins on the surface of the DED as a more favorable site of anchorage than the ppAAc surface so that, when the carrier is removed, the cells remain in contact with the DED.
Using eGFP-labeled cells, we were able to see by fluorescence microscopy that cells had penetrated into the DED after delivering them from our carriers. However, cells could only be seen to a depth of 100 μm or less (Fig. 6D), which corresponds to the depth of the contours in the DED's surface (Fig. 6G). Thus it is possible that the cells while well attached did not migrate into the dermis.
The reason for this may be that the basement membrane (as indicated by the presence of collagen IV which is a major constituent of it as shown in Fig. 6H) can be a barrier to their migration. Proteins of the basement membrane can be degraded by matrix metalloproteinases (MMPs); the principal one produced by fibroblasts and keratinocytes is MMP-9, and has a broad range of specificity, cleaving collagen IV, laminin, aggrecan, and elastin. 38
Cell types of mesodermal origin, such as fibroblasts, endothelial cells, and MSCs, constitutively express MMP-2 but only express MMP-9 upon stimulation.39–41 Dermal fibroblasts have been shown to heavily upregulate MMP-9 only upon interaction with keratinocytes as demonstrated in both transwell systems and direct coculture,42,43 thus providing evidence that communication between these two cell types is essential for modulating MMP production.
If MSCs behave similarly to fibroblasts in this respect, MMP-9 may only be produced when keratinocytes are present and MMP-9 levels may not be sufficient to promote basement membrane remodeling to allow for passage of MSCs into the dermis. The use of a more complex model with keratinocytes would be necessary to test this hypothesis.
The aim of this work was to demonstrate the transfer of cells from a carrier successfully to DED. Future studies requiring the use of more sophisticated in vitro wound models, where keratinocytes and fibroblasts and possibly endothelial cells are present, and also in vivo studies are now needed to assess whether MSCs transferred from our carriers actively contribute to wound healing.
An important feature of this study was that we cultured hBM-MSCs on the carriers for periods of up to 3 days and confirmed that their phenotype, viability, and ability to support vascular tubule formation were unaffected (Figs. 3 and 4). In addition cell transfer to DED after this time was evident (Fig. 5). This is a key finding because 3 days is a window long enough for carriers to be seeded with cells sourced from a stem cell bank and to be transported anywhere in Europe. Thus MSC treatment could be provided to patients who live a considerable distance from an accredited and licensed cell therapy laboratory. In addition, the MSCs could be prescreened by the bank for their ability to support vascular tubule formation, which will vary among donors based on age and other factors, and thus enhance their quality and potential to promote revascularization of wounds and more effective wound healing.
Current methods of MSC delivery to skin include intradermal injection around the wound site9,10,13 and the use of fibrin sprays. 8 Delivery by injection concentrates cells to a single locality with the possibility that cells will migrate from the site thereafter. However, this method may prove impractical for large areas of tissue loss because multiple injections may be required. In addition, creating a suspension of cells requires further cell manipulation and the use of trypsin, which can be potentially damaging to the cells. Cells delivered in matrices such as matrigels 9 or fibrin sprays 8 also have their limitations. Matrigel is not suitable for use in the clinic because these reconstituted basement membranes are extracted from mouse sarcoma cells and, therefore, would not pass the stringent European regulatory requirements. 30 Fibrin sprays may be composed of human fibrinogen and thrombin, but require complex preparation and have a short working time. 8 To the best of our knowledge there are no other cell carriers, derived from synthetic materials, used for the delivery of cells to skin, although it is possible that cell-sheet technology maybe applied to deliver cells to skin. For example, the thermoresponsive polymer poly(N-isopropyl)acrylamide has been used to create sheets of adipose-derived MSCs for myocardial infarction repair. 44 However, the authors found it necessary to transfer the cells to an elastic plastic sheet for handling, presumably because the original cell sheets were too difficult to handle due to their thin and fragile nature for application to the beating heart.
In light of the above, the chief advantage of our delivery approach is that it is a simple one-step method where no trypsin or temperature decrease is required. Coupled to the material's ease of handling and the potential to cover large skin defects, this method may be favored by clinicians for delivery of MSCs to wounds. There is keen interest in developing products for the clinic that are devoid of animal components and our carriers help to achieve this.
Another challenge for clinical uptake of MSC therapy is the development of a xenobiotic-free media. In the future it is possible that serum-free media 45 or media supplemented with human platelet lysate 46 will be used to culture the cells on the carrier. However, there are as yet no xenobiotic-free media approved for clinical use. Accordingly in these studies, we used bovine sera from approved sources such as Australia or New Zealand from herds free from bovine spongiform encephalitis, which have been approved for clinical use for many years. With any change of media in the future, it will be important to study how the MSCs respond to these media on the chemically defined surfaces as we have shown that the media composition can affect the culture of cells on ppAAc surfaces. 18
With the clinic in mind, other important factors to consider are the cost of scale-up of production and the long-term stability of the surfaces. It has been reliably demonstrated that ppAAc surfaces can be prepared to be stable for up to 300 days, 47 and with regards to scale-up, vacuum pressure technology is now widespread for preparing coatings in industry. 48
Conclusions
We have shown that synthetic cell carriers can be produced by plasma polymerization of acrylic acid onto medical-grade silicone, for the purpose of delivering hBM-MSCs to skin. The reliable surface chemistry enhanced the attachment of hBM-MSCs to the silicone substrate without affecting their phenotype, viability, and their functional ability as assessed by their support of vascular tubule formation. We also demonstrated the successful transfer of the cells to an in vitro skin model, and that cells remained viable on the dermis after leaving the carrier for at least 7 days. All of the above support the use of this synthetic carrier for clinical delivery of MSCs.
Footnotes
Acknowledgments
This work was supported by research funding from the EU Framework VII Cascade grant (G.T., S.M.W., A.M., S.M., P.C.E., and L.E.S.), NHS Blood and Transplant (A.M., G.T., and S.M.W.), National Institutes of Health Research (A.M., G.T., and S.M.W.), and Restore Burns and Wound Healing Trust (S.M.W.). This report presents independent research commissioned by the National Institute for Health Research (NIHR) under its Program Grants scheme. The views expressed in this publication are those of the author(s) and not necessarily those of the NHS, the NIHR, or the Department of Health.
Disclosure Statement
No competing financial interests exist.