
Abstract
Our long-term goal is to treat osteochondral lesions with bioengineered biphasic constructs. We have previously demonstrated that biphasic constructs, created in vitro with primary chondrocytes harvested from healthy joints and a porous calcium polyphosphate (CPP) substrate bone substitute, could successfully repair a focal defect in sheep joints. However, primary chondrocytes are limited in supply and cannot be used in engineering constructs large enough for clinical use. Thus, we developed a robust protocol to predifferentiate sheep bone marrow-derived stromal cells to chondrocytes on collagen-coated polytetrafluoroethane membrane inserts, and harvest the chondrocytes that develop and subsequently culturing these predifferentiated cells scaffold-free on the intended articulation surface of the CPP. Chondrocytes predifferentiated on membrane culture accumulated similar matrix as those in conventional pellet culture, but expressed less Col1a1 RNA. Membrane culture predifferentiated cells gave rise to a functionally superior hyaline cartilage tissue compared to pellet culture predifferentiated cells. Studies demonstrated that 2 weeks of membrane predifferentiation culture followed by 8 weeks of biphasic construct culture was the optimal culture period at which the compressive mechanical strength and the accumulation of extracellular matrix were maximized while avoiding tissue mineralization. This protocol will be used to generate implants for preclinical study to determine their ability to repair osteochondral lesions.
Introduction
We have previously developed a method to engineer a biological substitute for damaged cartilage by reforming cartilage tissue from chondrocytes in vitro on top of calcium polyphosphate (CPP), a porous bone substitute biomaterial with a high compressive strength that exhibits excellent osseointegrative properties as demonstrated in vivo.2,3 These tissue-engineered cartilage-CPP biphasic constructs could replace the osteochondral plugs currently used in mosaicplasty to treat focal joint defects, eliminating the need for harvesting the plugs from healthy joints. These constructs have been characterized, 4 and used successfully in an in vivo focal defect study in sheep 5 —a clinically relevant model, as their knees' biomechanics are similar to those of humans. 6 However, in those studies, the cartilage tissue on CPP was formed using autologous articular chondrocytes harvested from healthy cartilage. One major problem limiting the clinical application of bioengineered cartilage for joint repair is identifying a source of sufficient numbers of differentiated chondrocytes to form enough articular cartilage to repair the large defects that occur in patients. Chondrocytes de-differentiate when passaged (to expand cell number) even once in monolayer culture.7–9 A variety of approaches have been developed to circumvent this.10–18 Re-differentiation of passaged chondrocytes by culturing cells in a three-dimensional environment allows the cells to assume a spherical morphology14,18,19 but under these conditions the cartilage phenotype is not fully restored. Alternatively, human embryonic stem cells can be induced to differentiate into chondrocytes in the presence of growth factors such as BMP-6 or when grown on scaffolds, suggesting that they may be another potential source of cells for tissue engineering cartilage.20–24 However, these cells are allogeneic and thus may require immunosuppressants or other strategies to overcome histocompatibility barriers.
Mesenchymal stromal cells (MSCs) could be an alternative cell source: they can be readily obtained from autologous donors, expanded, and differentiated into chondrocytes in vitro using pellet culture or on a biomaterial.20,25 Deriving chondrocytes from bone marrow-derived MSCs (BMSCs) obviates the need to harvest healthy cartilage, and the rapid proliferation of BMSCs in vitro expedites the process for obtaining a large number of cells to create cartilage tissue.
Thus, the aim of this study was to engineer biphasic implants consisting of cartilage formed in vitro from sheep BMSCs36–39 on and integrated with the intended articulation surface of porous CPP substrates. We found that it was necessary to employ an intermediate step in which BMSCs were cultured on cell culture inserts 40 to induce MSC differentiation to chondrocytes before seeding on the CPP. The optimal in vitro protocol was determined to yield tissue with sufficient biochemical and biomechanical properties for use in biological repair.
Materials and Methods
Isolation and expansion of BMSCs
Bone marrow samples were aspirated from the humerus of male sheep into heparinzed Vacutainers (Becton Dickinson, Mississauga, ON, Canada). Aspirates were filtered through a cell strainer (70 μm pore size) (Becton Dickinson). The filtrate was centrifuged (300 g) for 25 min at 4°C, and the pellet was subjected to red blood cell lysis using a solution of 0.144 M ammonium chloride (Sigma-Aldrich, Oakville, ON, Canada) in 17 mM Tris-HCl buffer, pH 7.7. The nucleated cells were washed and plated on monolayer at a cellular density of 2.5×105 cells/cm2 in expansion media (XM) composed of minimum essential media α (Invitrogen, Burlington, ON, Canada), 10% fetal bovine serum (FBS; Wisent, Inc., St.-Bruno, QC, Canada), 1 mM sodium pyruvate, and 1×penicillin/streptomycin (Invitrogen). Nonadherent cells were discarded after 24 h. Media was changed every 3 days until the monolayer cultures were 80%–90% confluent, at which the cells were enzymatically harvested with 0.05% trypsin-EDTA (Invitrogen) and re-plated at a cell density of 5.0×103 cells/cm2 in XM. These cells were again cultured until 90% confluency was attained, followed by enzymatic harvesting by trypsin-EDTA, washing, and cryopreservation in 50% FBS, 40% XM, and 10% DMSO (Sigma-Aldrich) until later use.
Chondrogenic predifferentiation of BMSCs
Cryopreserved BMSCs were thawed, plated in monolayer at a cell density of 7.5×103 cells/cm2 in XM, and expanded to 90% confluence. The BMSCs were harvested and suspended in a defined chondrogenic media (CM) composed of high-glucose Dulbecco's modified Eagle medium (DMEM), 1×ITS cell culture supplement (BD Biosciences, Bedford, MA), 2 mM GlutaMAX (Invitrogen), 1 mM sodium pyruvate, 100 nM dexamethasone (Sigma-Aldrich), 100 μg/mL ascorbic acid 2-phosphate (Sigma-Aldrich), and 10 ng/mL transforming growth factor-β3 (R&D Systems, Minneapolis, MN). The MSC were then cultured either as pellet or membrane culture to induce chondrogenesis (predifferentiation step). For pellet cultures, 5×105 cells were centrifuged in wells of a 96-well round-bottom nontissue culture-treated polypropylene plate (Corning, Corning, NY) with 250 μL of CM and placed in culture in the incubator. At day 3, pellets were transferred to a 96-well tube rack and cultured in 500 μL of CM per tube. The medium was changed twice a week. For membrane cultures, 12-mm-diameter cell culture insert membranes (0.2 μm pore size; Millipore, Billerica, MA) were coated with human collagen type IV (Sigma-Aldrich) in 0.1 N acetic acid overnight. The membranes were then incubated at 37°C with 100 μL of FBS for 2 h. A 400 μL cell suspension containing 2.0×106 BMSCs was placed on the membrane, left for 3 h in the incubator before additional CM was added to a total volume of 2 mL. Medium changes were performed every 2–3 days and the cultures were grown for various times up to 3 weeks.
Cartilage-CPP biphasic construct culture
Cylindrical CPP rods of 4 mm diameter were prepared by gravity sintering 75 to 106 μm CPP powder particles in platinum containment tubes at 950°C as previously described, 4 and disks of 2 mm thickness were cut from these rods (Fig. 1). The disks were placed in Tygon tubing to create a well-like structure and subsequently γ-irradiated (2.5 MRad). The cartilage tissue generated by predifferentiation was digested in 0.5% w/v collagenase A (Roche Diagnostics, Indianapolis, IN) in F12 media with periodic agitation for 90 min at 37°C. The cells were washed twice, and then placed on the top surface of CPP disks (2×106 cells/disk in 30 μL). The biphasic constructs were cultured in DMEM-F12 (50:50) media supplemented with L-glutamine (1:1; Invitrogen) and 5% FBS, 1 mM sodium pyruvate, and 100 μg/mL ascorbic acid 2-phosphate. After day 4, the FBS concentration was increased to 20% and the media changed every other day. The tubing was removed at 1 week and the cultures harvested at either 4 or 8 weeks for analysis.

The calcium polyphosphate disks. Gravity sintering of calcium polyphosphate (CPP) powder yielded porous, biodegradable material on which bone marrow-derived stromal cell (BMSC)-derived cartilage was grown. Gross appearance
Histological and immunohistological evaluation
In vitro-formed tissue was removed from their substrates, either the membrane or the CPP, fixed in 10% neutral formalin buffer, and embedded in paraffin. Four-micrometer sections were cut and stained with hematoxylin and eosin, toluidine blue (pH 3.0), or von Kossa stain with fast red counterstain. For collagen type I immunostaining 41 paraffin-embedded sections were rehydrated and digested with 2.5 mg/mL trypsin and 25 mg/mL hyaluronidase, blocked with 20% goat serum, and incubated with antibody reactive with collagen type I (CalBioChem, La Jolla, CA) overnight at 4°C. Subsequently, samples were incubated with goat anti-mouse secondary antibody labeled with Alexa Fluor 488 fluorophore (Invitrogen) and counterstained with DAPI. Collagen type II immunostaining was carried out as previously described. 42 Paraffin-embedded sections were rehydrated and digested with 10 mg/mL pepsin for 6 min at 37°C, blocked with 2% (v/v) horse serum, and incubated with an antibody reactive to collagen type II (mouse monoclonal; Labvision, Fremont, CA). Immunoreactivity was detected using biotinylated horse anti-mouse secondary antibody (Vector Laboratories, Burlington, ON, Canada), Vectastain Elite ABC kit (Vector Laboratories) and diaminobenzidine with hematoxylin counterstain.
Biochemical analysis
Biochemical properties of tissues were assayed as previously described. 42 Briefly, tissues were detached from their substrates and snap-frozen at −80°C. Frozen samples were digested with 40 μg/mL papain (Sigma-Aldrich) in a buffer containing 20 mM ammonium acetate, 1 mM EDTA, and 1 μM DTT for 48 h at 65°C and stored at −30°C until further analysis. The DNA content of the digest was determined by using the Hoechst 33258 dye and fluorometry with the emission wavelength of 458 nm and the excitation wavelength of 365 nm. A standard curve was generated using calf thymus DNA (Sigma-Aldrich) in PBS. The proteoglycan content of the digest was estimated by quantifying the amount of sulfated glycosaminoglycans (GAGs), using the dimethylmethlene blue dye and spectrophotometry with a wavelength of 525 nm. The standard curve for the proteoglycan content assay was generated using chondroitin sulfate (Sigma-Aldrich). Collagen content of the digest was estimated by quantifying the hydroxyproline content after acid hydrolysis at 110°C using chloramine-T/Ehrlich's reagent assay and spectrophotometry with a wavelength of 561 nm. The standard curve for the collagen content was generated using hydroxyproline (Sigma-Aldrich). To calculate the collagen content it was assumed that hydroxyproline comprises approximately 10% of the weight of collagen.
Gene expression analysis
Tissues were homogenized by glass bead milling (Cole Parmer, Laval, QC, Canada). Total RNA was isolated using the Nucleospin II RNA isolation kit (Mackerey-Nagel, Duren, Germany), treated with DNase (Ambion, Austin, TX), and quantified with Nanodrop (ThermoFisher, Wilmington, DE). cDNA was synthesized from 500 ng of total RNA using SuperScript II (Invitrogen) and random hexamers. Quantitative PCR (qPCR) was performed using Roche LightCycler 480 (Roche Diagnostics) and a standard protocol in a reaction volume of 10 μL containing SYBR master mix (Roche Diagnostics) and 0.5 μM primers. 43 Standard curves were generated using serial dilution of cDNA to determine the efficiency of each primer pair. Products of qPCR primers were sequenced to verify the amplification of intended gene targets.
Stress relaxation assay for compressive modulus
The compressive modulus of cartilaginous tissue of the biphasic construct was determined using stress relaxation testing and the Mach-1 mechanical testing apparatus (BioMomentum, Laval, QC, Canada) with a 0.65-mm-diameter indenter as previously described. 5 The thickness of the cartilage tissue was estimated by measuring the thickness from the lateral aspect of the construct with a calliper. At each step, 1% strain was applied while allowing unconstrained lateral deformation, and the compressive force was allowed to relax until an equilibrium force level, defined as a change of 2 dynes/min, was reached. This was repeated 20 times, and the compressive modulus was estimated from the best-fit linear regression of stress–strain relationship containing at least 10 data points.
Statistical analysis
Two-way analysis of variance (ANOVA) was used to analyze the effects of culture conditions and variance among donor animals. Unbalanced two-way ANOVA with general linear tests was employed where appropriate. In all cases, outcome was attributed much more strongly to variation in culture condition than to variation among animals. Hence, biochemical and biomechanical data from various CPP culture conditions were evaluated using one-way ANOVA and Tukey post hoc testing. Significance was assigned at p<0.05.
Results
Chondrogenic predifferentiation of BMSCs in membrane and pellet cultures
We first compared the effectiveness of membrane and pellet culture differentiation of sheep BMSCs to chondrocytes. Histological examination of 2- and 3-week cultures revealed cartilage tissues rich in extracellular matrix, the bulk of which contained cells with round morphology, whereas peripheral portions of the tissues interfacing the culture media contained cells with more flattened morphology (Fig. 2). In membrane cultures, the distribution of cells and extracellular matrix appeared uniform through its thickness. Toluidine blue staining showed that the accumulated matrix was rich in sulfated proteoglycan, although less staining was observed at 2 weeks than 3 weeks of culture (Fig. 2E, F). The pellet cultures yielded a heterogeneous sphere of matrix-rich tissue in which the cells had a round morphology. However, in contrast to the membrane cultures, there was a distinct thin outer layer that was more cellular and showed weaker staining with toluidine blue, suggesting that these cells were less chondrocytic (Fig. 2C, D).

Histological appearance of tissue derived from membrane (left) and pellet cultures (right) of BMSCs cultured for 2 and 3 weeks in defined chondrogenic media. Hematoxylin & eosin staining
The accumulation of proteoglycan and total collagen in membrane and pellet cultures was quantified over the 3-week period (Fig. 3). By 3 weeks, the amount of GAG and collagen in the tissues were similar in both pellet and membrane cultures when normalized to their respective DNA content. However, while membrane cultures exhibited a steadily increasing accumulation of GAG and collagen over the 3-week period, the pellet cultures exhibited a more rapid increase that reached its maximum by 2 weeks of culture and then decreased (3 weeks).

Accumulation of extracellular matrix in tissue derived from membrane and pellet cultures of BMSCs cultured for up to 3 weeks in defined chondrogenic media. Sulfated proteoglycan
Transcript levels of chondrogenic markers were assayed over a 3-week period using qPCR (Fig. 4). Cells in membrane culture expressed a significantly lower level of Col1a1 than those in pellet cultures from day 7 onward (Fig. 4A). Meanwhile, expression levels of Col2a1 and aggrecan (Agc1; Fig. 4B–D) increased over time up to 2 weeks in culture in both pellet and membrane culture, after which time expression levels of both began to decrease. Sox9 expression showed a biphasic peak in both cultures with levels significantly higher in pellet cultures. Expression levels of chondrocyte hypertrophic markers, Runx2 and Col10a1, also increased in both culture types, suggesting that the method of culture did not significantly alter their potential to terminally differentiate as previously observed.
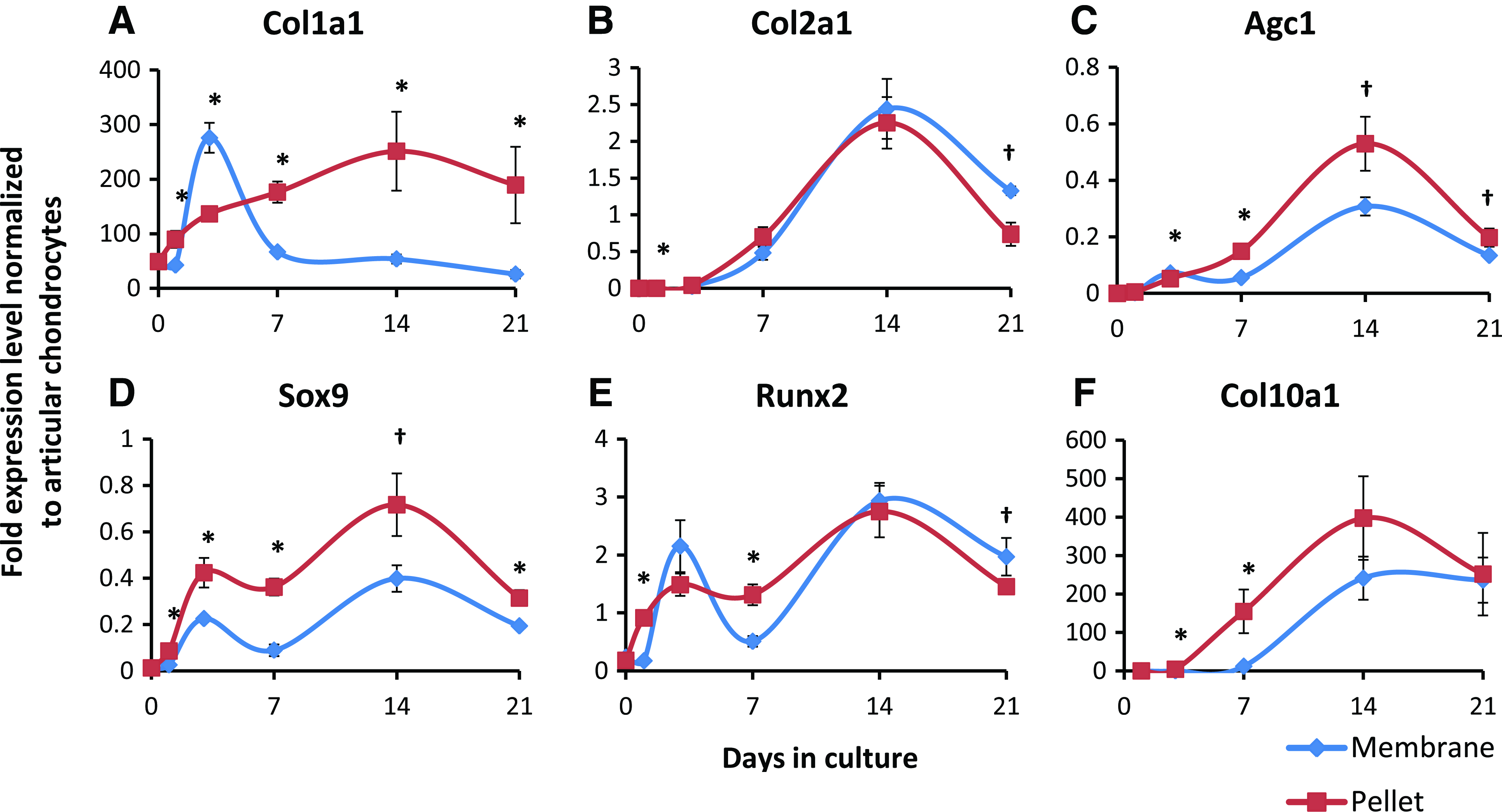
Gene expression of chondrogenic markers by tissue derived from membrane or pellet cultures of BMSCs cultured for up to 3 weeks in defined chondrogenic media. Transcript levels were assayed by qPCR with 18s rRNA as the reference gene and compared to those of native ovine chondrocytes. Cells in membrane culture expressed a lower level of Col1a1
Predifferentiated cells from membrane cultures form better cartilage tissue in biphasic constructs
At first, BMSCs were seeded directly on the CPP substrate and cultured in CM to create the cartilage-CPP biphasic construct; however, this did not result in cartilage tissue formation (data not shown). Therefore, we investigated whether cells that were “predifferentiated” in membrane and pellet cultures could form tissue on the CPP substrate. After 3 weeks of differentiating sheep BMSCs in pellet and membrane cultures to chondrocytes, tissues were digested using collagenase to isolate the predifferentiated cells. The cells isolated from pellet and membrane cultures were then cultured scaffold-free on the CPP substrate for 4 weeks and the resulting tissues characterized (Fig. 5). Histological examination showed that cartilage tissues formed by membrane predifferentiated cells yielded were comparable in thickness to those formed by pellet predifferentiated cells, but accumulated more proteoglycans and collagen type II, and less collagen type I, than the pellet predifferentiated counterpart (Fig. 5A). However, when normalized by cellularity as reflected by the tissues' DNA content, the difference in proteoglycan accumulation between the two types of tissues was not statistically significant (Fig. 5B), while tissues formed by pellet predifferentiated cells accumulated significantly more total collagen (Fig. 5C). Stress relaxation testing showed that the tissue formed by membrane predifferentiated cells had a significantly higher compressive modulus (Fig. 5D). This, combined with the more desirable accumulation pattern of collagen, suggested that chondrocytes predifferentiated from BMSCs in membrane culture was best suited for creating cartilage-CPP biphasic constructs than either the undifferentiated BMSCs or pellet culture-differentiated chondrocytes.
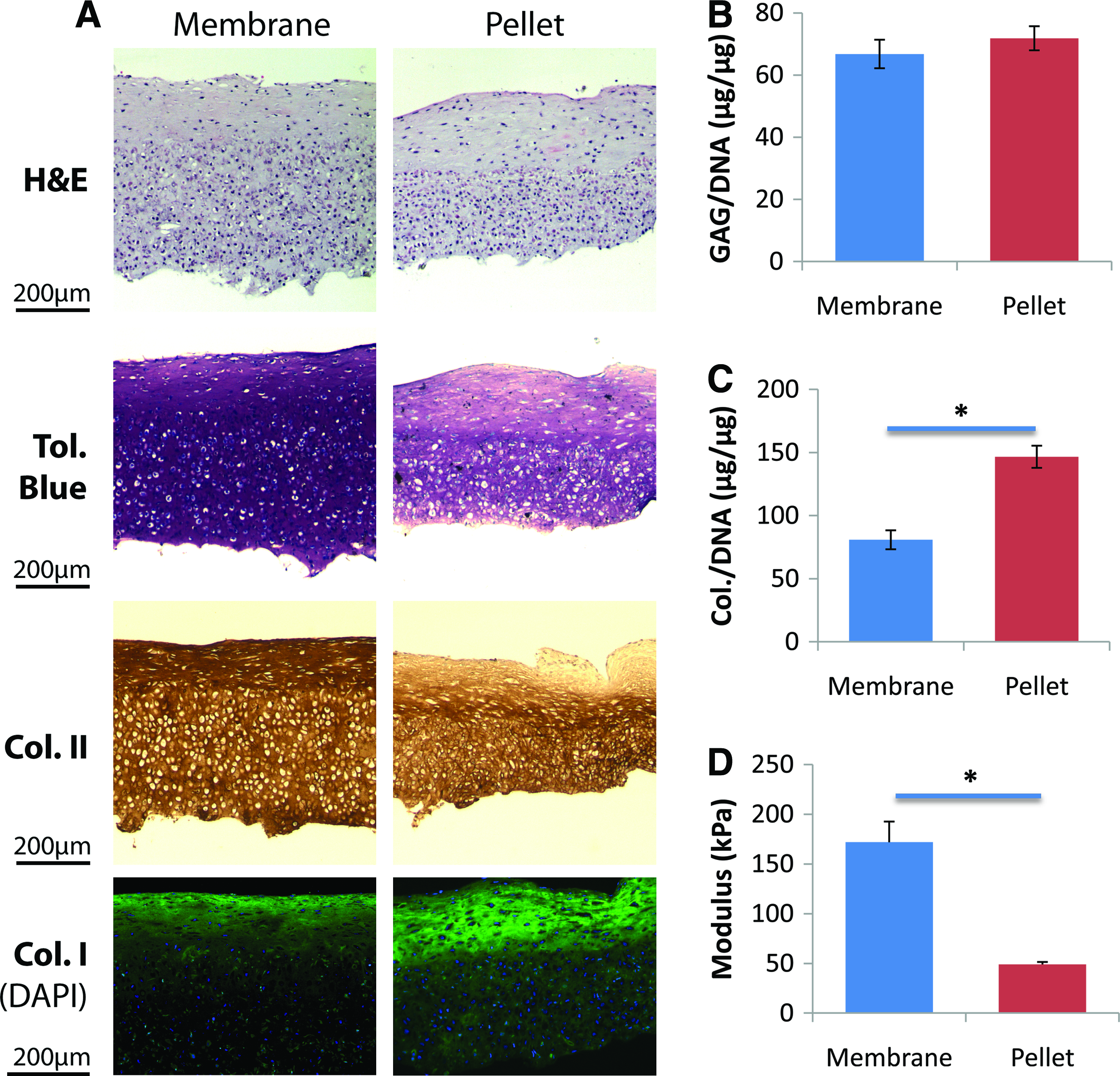
BMSCs predifferentiated in membrane culture yielded better cartilage on CPP than those predifferentiated in pellet culture. Cells were differentiated to chondrocytes in membrane or pellet cultures, enzymatically isolated, and cultured on the CPP substrate for 4 weeks. Histology of the tissues on CPP
Optimization of biphasic construct tissue culture protocol
To optimize the membrane culture protocol for creating biphasic constructs, we first varied the duration of predifferentiation membrane culture and histologically examined the resulting cartilage tissue formed on the CPP substrate after 3 weeks of culture. BMSCs predifferentiated for 1 week formed a thin, fibrous layer of tissue with spindle-like cell morphology, indicating the absence of cartilage tissue (Fig. 6A). However, BMSCs predifferentiated for 2 or 3 weeks formed thick tissues rich in extracellular matrix containing cells with round morphology, suggesting that these tissues were cartilaginous (Fig. 6B, C, data not shown).
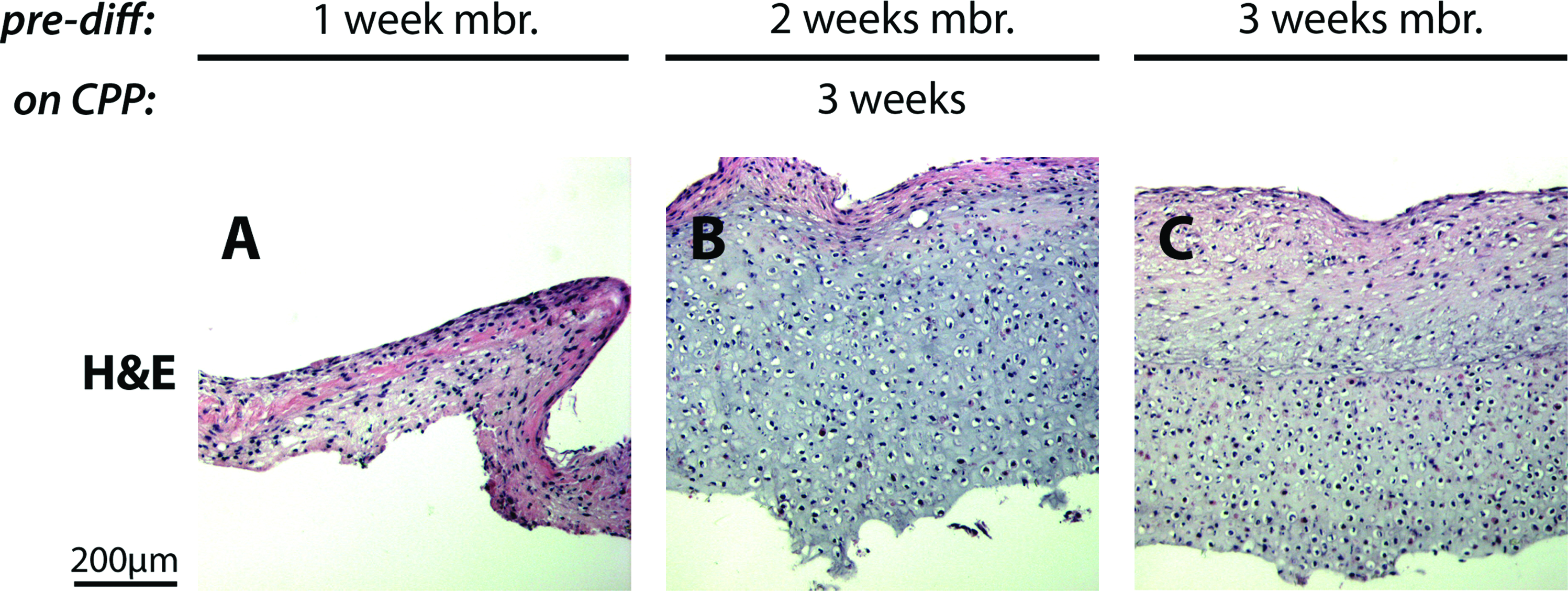
BMSCs must be predifferentiated for at least 2 weeks in membrane culture to form tissue on CPP after isolation. BMSCs were predifferentiated in membrane cultures for various lengths of time, enzymatically isolated and cultured on the CPP substrate for 3 weeks. BMSCs predifferentiated for 1 week
Subsequently, we varied the duration of post-differentiation culture on CPP and characterized the cartilage tissue histologically, biochemically, and mechanically. As the difference between tissues produced by 2-week and 3-week predifferentiated BMSCs was not clear, use of cells from both predifferentiation conditions were also explored. All culture combinations yielded tissues that accumulated extracellular matrix rich in proteoglycan and collagen, which appeared to increase in thickness with time in culture (Fig. 7A). Within the tissue, two layers could be observed: a top layer sparsely populated with cells and exhibiting weaker toluidine blue and stronger collagen type II staining, whereas the bottom layer was densely populated with cells and exhibited strong toluidine blue and weaker collagen type II staining. Of the different conditions, tissues derived from cells predifferentiated for 2 weeks and cultured on CPP for 8 weeks (Fig. 7A, second column) appeared to be the best condition because the least accumulation of collagen type I was observed among them (Fig. 7A, third row). The cartilage tissues cultured under this condition also had the highest proteoglycan and total collagen accumulation (Fig. 7C, D), as well as the highest compressive elastic modulus and tissue thickness (Fig. 7E, F), even when compared with the tissues formed by pellet predifferentiated cells (Fig. 5B–D, data not shown). In particular, the thickness of the cartilage tissues cultured under this condition approached the thickness of sheep native cartilage measured from the femoral condyle (Fig. 7F; p=0.032). Given the long in vitro culture period and the expression of Runx2 and Col10a1 in predifferentiation cultures (Fig. 4E, F), we confirmed that mineralization, a consequence of terminal differentiation, 45 did not occur at the end of the 8-week culture period, by staining the tissues with Von Kossa, as no mineralization was detected (Fig. 7B).
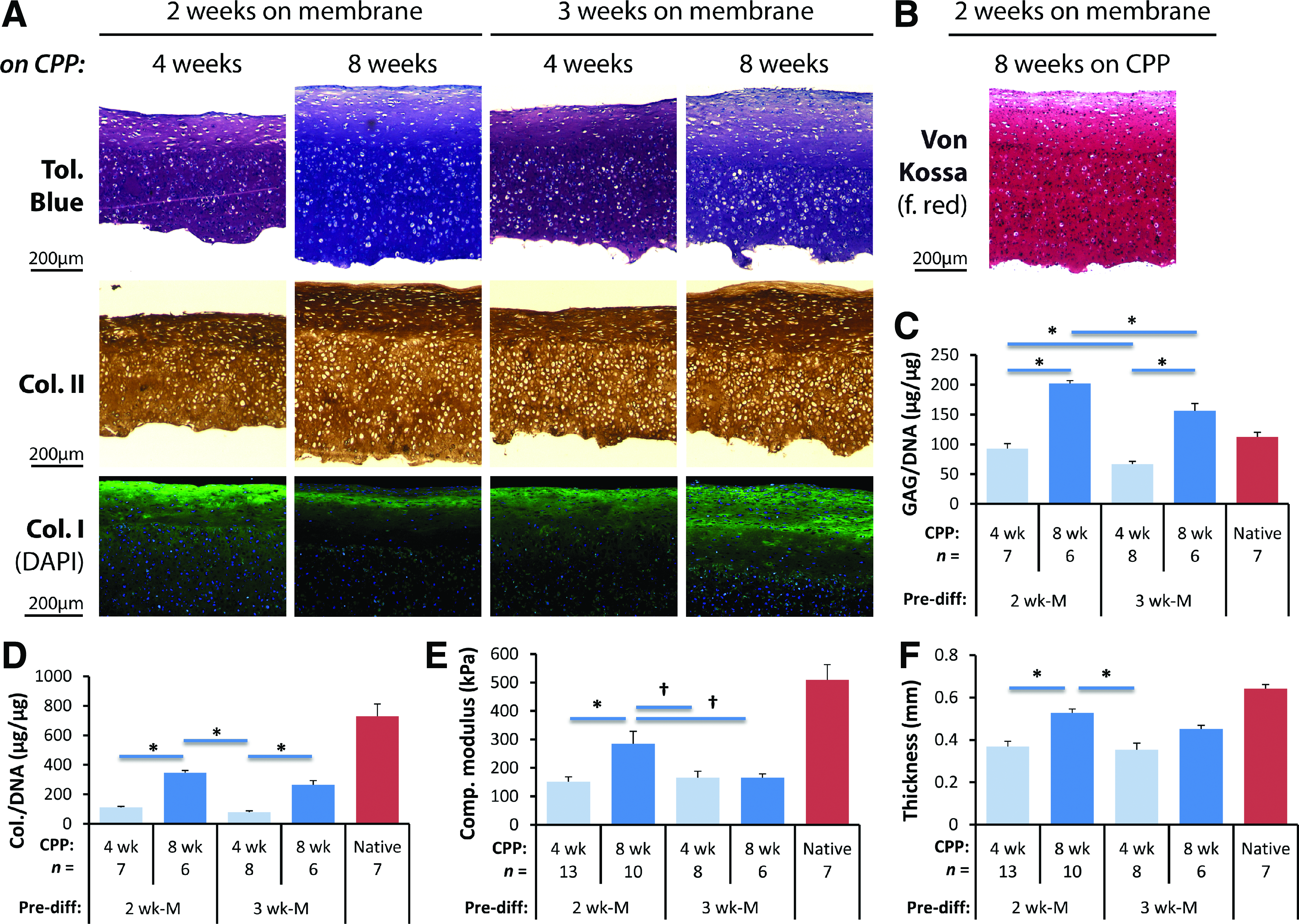
Optimal cartilage tissue was obtained by predifferentiating the BMSCs in membrane culture for 2 weeks and culturing the differentiated cells on CPP for 8 weeks. To optimize the culture conditions, BMSCs were predifferentiated on membranes for 2 or 3 weeks, cells isolated and cultured on CPP for 4 or 8 weeks. Histology
Discussion
We have successfully engineered cartilage-CPP biphasic constructs in vitro using sheep BMSCs by predifferentiating them to chondrocytes in high-density membrane culture. The cells predifferentiated in membrane cultures yield better cartilaginous tissue on CPP than those grown in conventional pellet cultures, and the length of time in predifferentiation and CPP culture affects the quality of cartilage tissue formed on the CPP substrate. Accumulation of extracellular matrix and the resulting compressive mechanical strength were maximized when BMSCs were predifferentiated in membrane culture for 2 weeks, isolated, and then cultured on CPP for an additional 8 weeks.
We noted that although membrane cultures and pellet cultures of BMSCs both yielded tissues with comparable matrix accumulation and gene expression profiles by 3 weeks of culture, the predifferentiated chondrocytes isolated from these tissues subsequently formed different cartilage tissues on the CPP substrate under the same culture condition. Chondrogenesis of BMSCs are conventionally carried out in micromass or pellet cultures to mimic in vitro the cellular microenvironment of condensed mesenchyme in skeletogenesis.26–28 While previous studies comparing alternate culture methods to conventional pellet culture35,40 contrasted the gene expression of the cells and the matrix they accumulated while being differentiated in different culture methods, we performed an additional step to isolate these cells and compare the cartilaginous matrix they accumulate in a subsequent culture. In doing so, we observed that predifferentiated cells isolated from the membrane culture formed a more collagen type II-rich, mechanically competent cartilage tissue than predifferentiated cells from the pellet culture: this was an unexpected observation, given that we found the tissues formed by differentiating BMSCs in membrane cultures and pellet cultures were comparable.
The cartilage tissue generated with our BMSC-derived chondrocytes did not mineralize despite the extended length of culture. Although BMSCs are well known for their chondrogenic potential, their tendency to mineralize has limited their use clinically for joint repair.44,46 In our protocol, no mineralized tissue developed even though cells were cultured using predifferentiated cells on CPP for up to 8 weeks in serum-supplemented media in the absence of pro-differentiation growth factors. This is not likely to be a culture artefact as previous work in our laboratory showed that deep-zone chondrocytes cultured on CPP will form a zone of calcified cartilage. 47 Further, mineralization can be induced in the tissues formed by predifferentiated BMSCs on CPP under the conditions described in this study if β-glycerophosphate is added to the culture media (data not shown). This is consistent with the in vitro hypertrophy model previously described. 48 Importantly when tissue-engineered cartilage formed by BMSCs was grafted orthotopically, Zscharnack et al. showed that these tissues do not undergo mineralization, suggesting that if the tissue does not mineralize in vitro it is unlikely to mineralize in vivo. 49
Although there still exists a disparity between the quality of the tissue formed in vitro with the currently developed protocol and the native articular cartilage, we have previously demonstrated that when placed in the in vivo environment, the biochemical and biomechanical properties of tissue-engineered cartilage will be improved. 5 This suggests that implantation of cartilage with properties less robust than native cartilage may still be sufficient; however, in vivo analyses will be required to confirm this. In contrast to scaffold-based cartilage tissue engineering strategies,29,50,51 the cartilage of the construct is free of artificial scaffolds as the tissue was formed by culturing a layer of cells on top of the CPP substrate.
Interestingly, while attempts to induce chondrogenesis of BMSCs directly on the CPP substrate failed, BMSCs predifferentiated to chondrocytes using either culture methods could readily establish tissues, though of different quality. The microenvironment of the substrate has a profound influence on the cells as has been shown for chondrocytes grown on biomaterials.45,52,53 During culture, the CPP substrate is known to release polyphosphates into the culture as it degrades hydrolytically. 3 It is not yet clear how they would affect the cells. It has been suggested that undifferentiated MSCs exposed to polyphosphates can influence the differentiation process in vitro. 54 Further, it may be that this release inhibits cartilage mineralization as we have shown previously in cartilage formed by deep-zone chondrocytes. 47
In summary, a method was developed that used BMSC-derived chondrocytes to substitute for articular chondrocytes in engineering a cartilage-CPP biphasic construct. This approach eliminates one of the limitations preventing clinical application of biphasic constructs. Preclinical studies are required to determine the efficacy of these constructs to repair osteochondral lesions.
Footnotes
Acknowledgments
We thank Dr. Bob Pilliar for preparing the CPP substrates and also Dr. Jian Wang and Cheryl Cui for technical assistance. The research was supported by a CIHR Team grant to R.A.K. and a CIHR operating grant to W.L.S. (FRN 62788). W.D.L. is a recipient of the Ontario Graduate Scholarship and the Ontario Graduate Scholarship for Science and Technology. W.L.S. is supported by a Canadian Research Chair.
Disclosure Statement
No competing financial interests exist.