
Abstract
Provision of a safe and secure supply of transfusible red blood cells (RBC) is a major global health challenge, and it has been proposed that manufactured RBC could help to alleviate the constraints of the current donor system. Several substantial challenges must be addressed for this approach to be feasible. At the most basic level, this relates to the large quantities of cells that are required: is there sufficient biological capacity, and is it possible to produce RBC using large-scale processes? While it has been demonstrated that, in principle, up to 5 units of RBC could be generated from a single donation of umbilical cord blood (UCB) hematopoietic stem cells, such yields are insufficient to supply demand and existing culture methods are unsuitable for large-scale manufacture. Given the capacity of the hematopoietic system in vivo, we reasoned that an optimized process should give rise to much larger quantities of RBC than previously reported. We successfully developed a robust ultra-high-yield RBC expansion process capable of producing over 500 units of RBC per UCB donation using fully defined culture medium. We obtained near-pure populations of reticulocytes with an enucleation frequency of >90%, mean cell hemoglobin content of 30.8 pg/cell, and mean cell volume of 133 fL. We also show that RBC can be efficiently produced in agitated bioreactor systems, demonstrating that no fundamental barriers exist to the manufacture of RBC using large-scale approaches.
Introduction
In 2005, Giarratana et al. 3 demonstrated that it is indeed possible to produce functional human RBC in vitro. Umbilical cord blood (UCB)-derived CD34+ hematopoietic stem and progenitor cells (HPC) were cultivated on feeder layers of murine MS5 cells or human mesenchymal stem cells in the presence of erythropoietin (EPO), stem cell factor (SCF), interleukin 3 (IL-3), and hydrocortisone (HC). Over a period of 21 days, numerical expansions of 1.95×106-fold (equivalent to ∼5 units of RBC per UCB donation) were reported for an immature erythroid cell population. Enucleation rates of 71%–91% were subsequently achieved by removal of SCF and HC from the medium, and continued cultivation on feeder-layers. Functionally, these cells were very similar to donor RBC and persisted in vivo to the same extent as native RBC. 3
While this approach demonstrated that unit quantities of functional RBC can be produced from HPC, the yields obtained are insufficient to supply demand and the process itself is unsuitable for large-scale manufacture of a transfusion product. In the United States alone 15 million units of donor RBC are transfused annually, 1 and the method described would require approximately two tennis courts worth of cell culture surface area to produce just one unit of RBC. 4 While several groups have subsequently attempted to eliminate the use of feeder layers,5,6 improve cell yields, 7 and shift to agitated suspension culture, 6 the method of Giarratana and colleagues 3 remains the most robust and efficient reported to date.
Given the high capacity of the hematopoietic system in vivo, we believe that the comparatively low cell yields reported so far reflect limitations in the culture process, not an intrinsic biological limitation of the HPC source. Indeed, Leberbauer et al. 8 achieved 108–109-fold expansion of erythroid progenitors in feeder-free culture. However, hemoglobin levels were ∼40% of donor levels and only limited enucleation was observed.
In this article we address the translation of laboratory flask-based RBC culture methods to an agitated bioreactor system, and describe an ultra-high-yield process capable of delivering 500 units of RBC per UCB donation (>108-fold numerical expansion).
Experimental Procedures
Cell culture
UCB donations were obtained with informed consent following institutional ethics review and approval. Within 24 h of collection, whole UCB was separated by density gradient centrifugation on Ficoll-Paque Plus (GE Healthcare Life Sciences), and the mononuclear cell fraction collected for magnetic isolation of CD34+ cells by MACS (Miltenyi Biotech) according to the manufacturer's instructions. Isolated CD34+ cells (fresh or frozen, >90% CD34+) were seeded at a density of 1×104 viable cells/mL into IMDM (Invitrogen) supplemented with 100 ng/mL SCF (Amgen), 3 U/mL EPO (Janssen-Cilag), 5 ng/mL IL-3 (ProSpec-Tany), and 10–3 M HC (Sigma-Aldrich), 1% fatty acid free bovine serum albumin (BSA; Sigma-Aldrich), 120 μg/mL holo-transferrin (Sigma-Aldrich), 10 μg/mL insulin (Sigma-Aldrich), 900 ng/mL ferrous sulfate (Sigma-Aldrich), and 90 ng/mL ferric nitrate (Sigma-Aldrich). On day 4, cultures were diluted 1 in 5 in fresh medium, and on day 8 fed again with fresh medium to give a cell density of 5×104 cells/mL. Thereafter, cultures were dilution fed to 1×105 cells/mL with fresh medium at regular intervals. Terminal differentiation and enucleation were induced by removal of SCF and HC.
After terminal differentiation, enucleated cells were obtained by filtration of the cell suspension using a PureCell Select Cell Harvest Kit (Pall Corporation). The cell suspension was passed directly over the filter unit, and the filtrate collected in to 50 mL tubes.
For GlutaMAX™ (Invitrogen) containing medium, maximum expansion was achieved in a 7% CO2 in air atmosphere. For cultures supplemented with L-Glutamine, a 5% CO2 in air atmosphere gave maximum expansion. All cultures were maintained at 37°C using humidified cell culture incubators.
Bioreactor culture
Cultures were initiated and treated as above until day 8. After feeding to give a cell density of 1×105 cells/mL in a minimum volume of 200 mL, cultures were then transferred to a 1 L CultiBag RM and mounted on a BIOSTAT CultiBag RM bioreactor system (Sartorius-Stedim biotech S.A.). Cultures were agitated at 15 rocks per minute at an angle of 8°, with a 50% N2 and 5% CO2 in air mix supplied at 0.2 liters per minute. At regular intervals, cultures were demi-depleted and fed with fresh medium to give a final cell density of 1×105 cells/mL in a minimum volume of 200 mL. In the later stages of culture, total volume was progressively increased to 1 L by dilution feeding.
Characterization
Cells were blocked with anti-FcR (Miltenyi Biotech) and BSA, and incubated with anti CD235a-APC and anti CD71-FITC antibodies (Biolegend) for analysis by flow cytometry. Population frequencies were determined relative to unstained autofluorescence controls.
Cell volume was determined using a Quanta MPL (Beckman Coulter). Mean cell hemoglobin was determined using a QuantiChrom Heme Assay Kit (BioAssay Systems) as per the manufacturer's instructions. Enucleation frequencies were determined from cytospin preparations stained with Leishmans stain, and between 100 and 200 cells/culture were classified. The hemoglobin composition of ex vivo expanded RBCs was determined using Variant II (BIO-RAD) hemoglobin analyzer by local medical pathology services.
Results
Feeder-free expansion of enucleated RBC
Due to the complexity of establishing and maintaining co-cultures of mixed cell populations, a first step in the development of a robust and scalable process for RBC manufacture was to eliminate the need for feeder cells. Using a simple dilution feeding regime and medium supplemented with the cytokines SCF, EPO, and IL-3 to drive proliferation and erythroid differentiation of UCB CD34+ HPC, we were able to generate RBC with similar yields to those reported by Giarratana et al. 3 After 21 days of culture, our feeder-free protocol achieved a log-mean increase in cell number of 1.73×106-fold (range=9.48×105–1.35×107; n=15; Fig. 1A). The expanded cell population exhibited a pure erythroid phenotype of mixed maturity (Fig. 1B). Consistent with the findings of Giarratana et al., 3 when these expanded cells were subsequently cultured in medium absent of SCF and HC (and feeder cells), nuclei were extruded and could be observed as independent bodies by fluorescent microscopy (Fig. 2A). Enucleation frequencies of >90% were obtained (Fig. 2B).
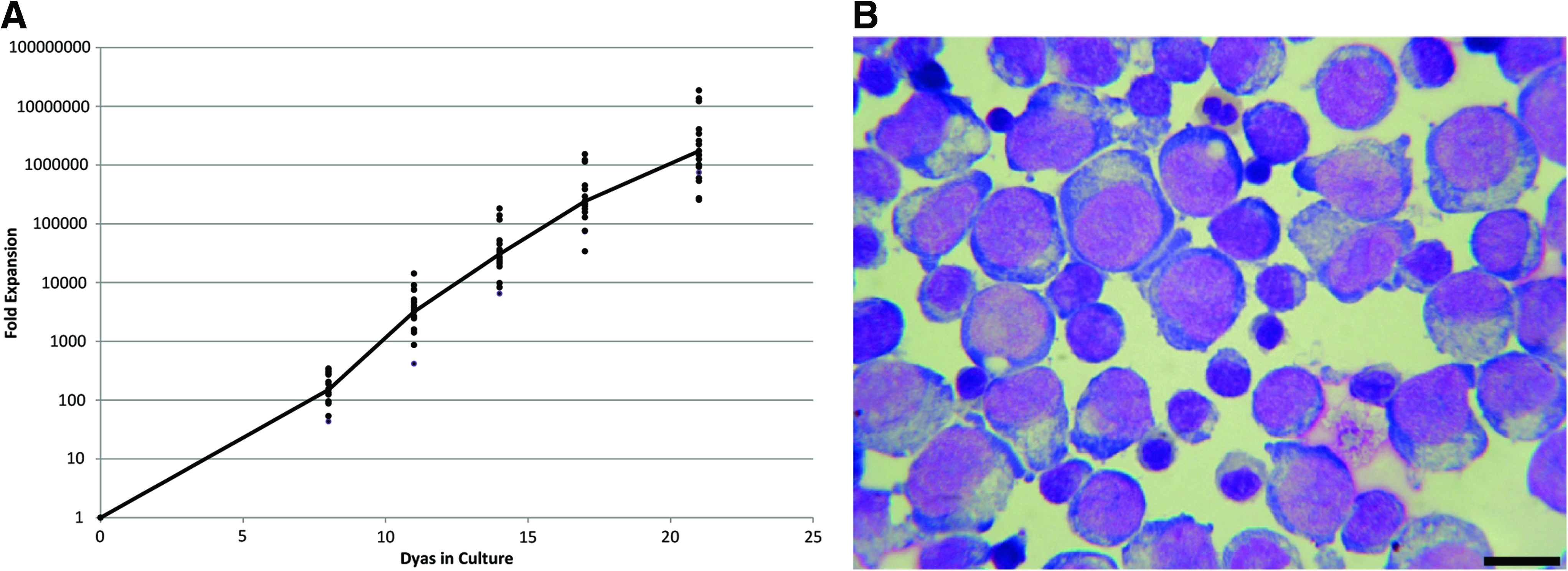
Expansion of erythroid cells in feeder-free culture.
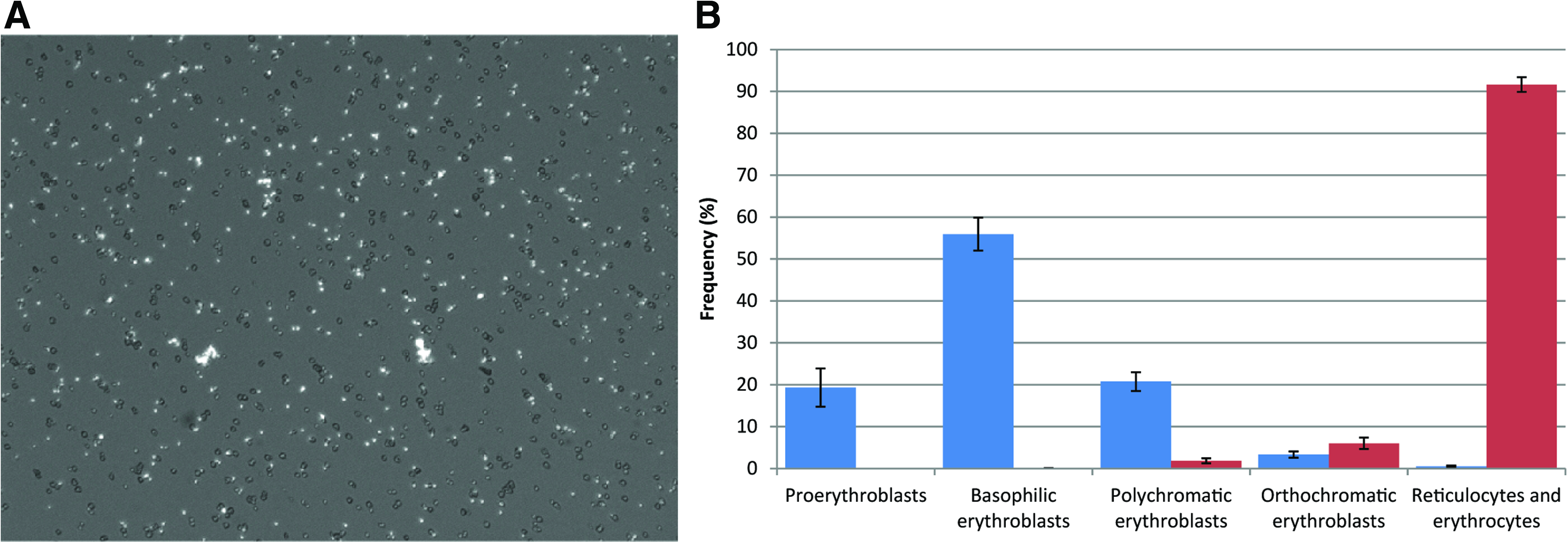
Enucleation of expanded erythroid cells in feeder-free conditions.
RBC culture in agitated bioreactors
Our feeder-free protocol relies entirely on suspension culture, and translation to a scalable bioreactor system is relatively straight forward. Adapting a process initially developed for clinical scale manufacture of neutrophils from HPC, 9 we first maintained low cell density cultures under static conditions for 8 days, and subsequently transferred to a wave-type CultiBag bioreactor. In our first attempt, expansion in 1 L bioreactor cultures was only 33% of the static control, and the cells exhibited a more mature phenotype (data not shown). We suspected this may be due to accelerated differentiation in response to high dissolved oxygen (DO) levels. In a subsequent bioreactor culture where DO was controlled to 50%, expansion was similar to that for static controls (Fig. 3). Thus, we were able to maintain expansion potential in agitated culture, enabling initial process scale-up to a volume of 1 L.
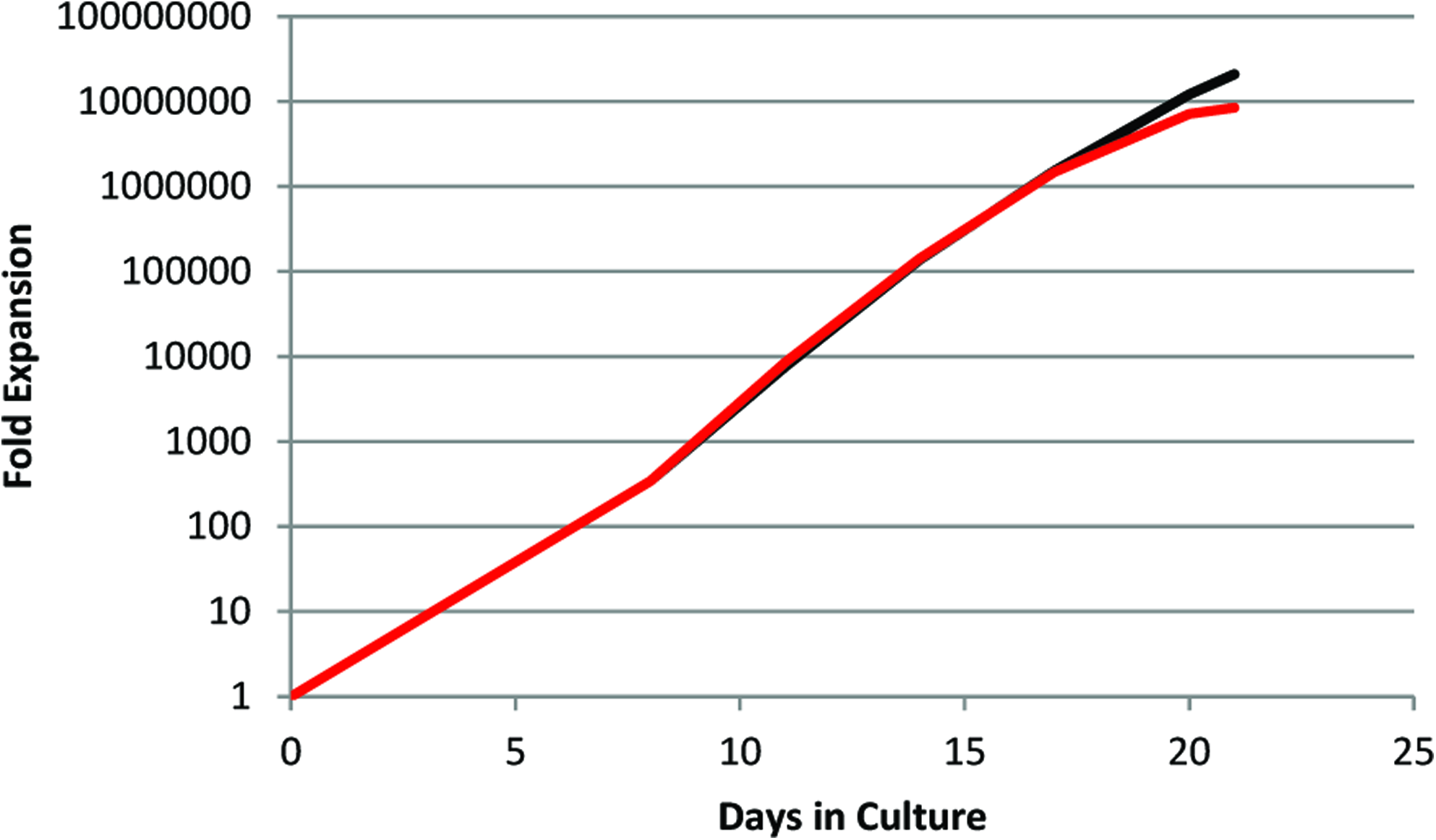
Erythroid cell expansion of CD34+ cells in 1 L bioreactor culture (50% dissolved oxygen, red line) and static control (black line). Color images available online at
Ultra-high-yield manufacture of RBC
While our cell yields were similar to those reported by Giarratana et al. 3 over the same culture duration, it was clear that in our more defined feeder-free conditions, cultures were still actively growing at day 21 (Fig. 1A). We reasoned that by extending the duration of expansion culture, RBC yields could be improved. When cultures were continued to day 33, a log-mean expansion of 2.25×108-fold was obtained (range 3.93×107–6.94×108; n=11; Fig. 4). This corresponds to a 130-fold improvement in cell yield, or ∼560 units of RBC per UCB donation (assuming 5×106 CD34+ cells/donation 3 ). While it was possible to further increase cell yields by again increasing culture duration, terminal differentiation was less robust and the red coloration typical of well-hemoglobinized cells was lost (data not shown).

Expansion of erythroid cells in extended duration cultures. By day 33, a 2.25×108-fold log-mean expansion in cell number was obtained (n=11).
Product characterization
Over the course of expansion we observed an expected decrease in expression of the transferrin receptor CD71 and sustained expression of glycophorin A (CD235a) (Fig. 5A, B). After maturation of the expanded erythroid progenitor cells in the absence of SCF and HC, cultures were passed through a PureCell Select Cell Harvest Kit (Pall Corporation) to remove free nuclei and remaining nucleated cells. Filtrates comprised near-pure populations of enucleated reticulocytes (Fig. 5B) with a median cell volume of 133 fL (Fig. 5C). The measured volume is consistent with that of young donor reticulocytes as reported by Gifford et al. 10 The mean hemoglobin content of manufactured cells was 30.8±0.5 pg/cell (n=3) compared to 27–33 pg/cell for normal donor RBC, with a mean composition of 50.8%±10% adult and 47.3%±7% fetal hemoglobin (n=3).
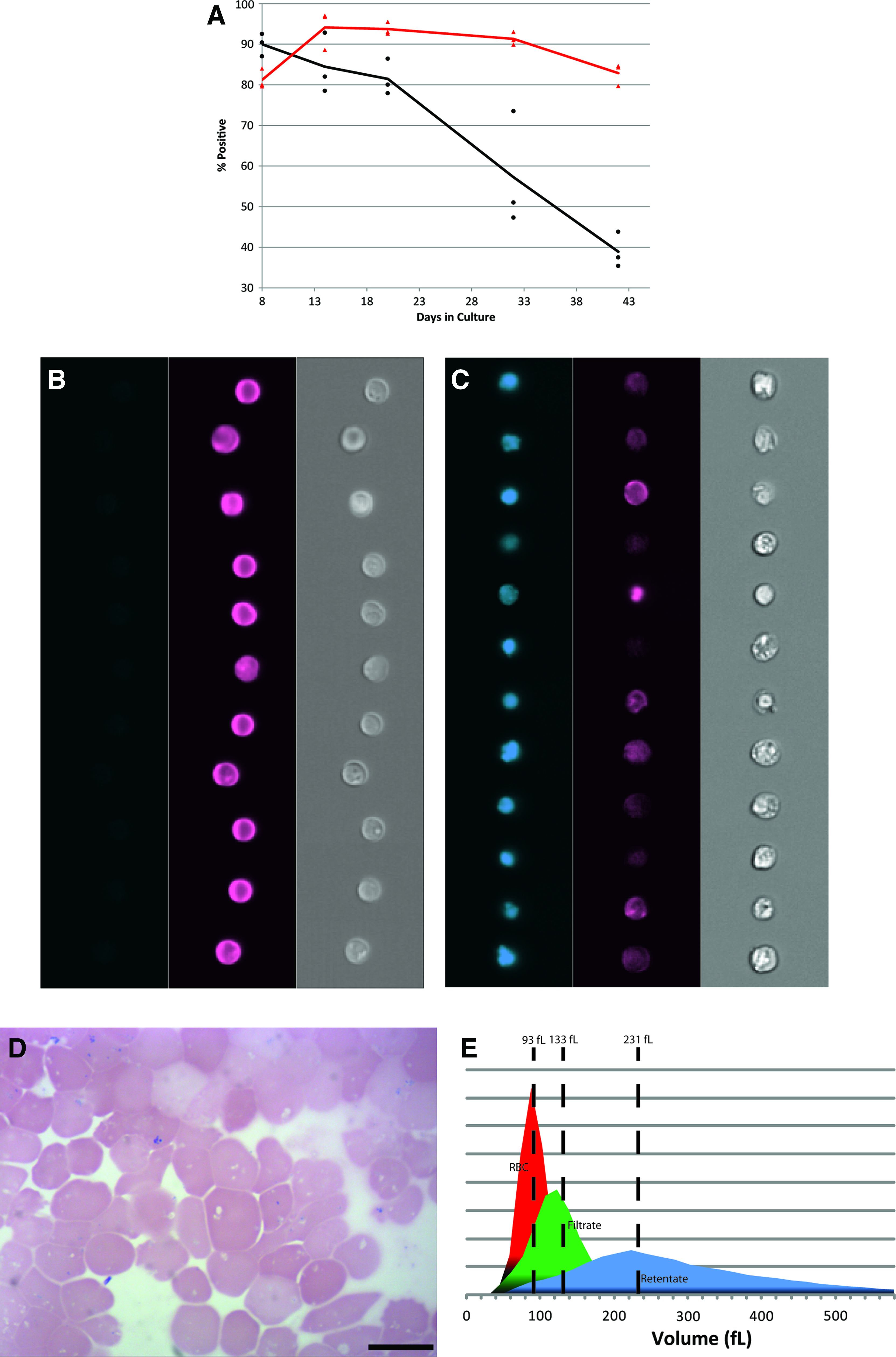
Characterization of erythroid cells generated in long-term cultures.
RBC manufacture with fully defined animal component-free medium
To reduce the possibility of disease transmission and increase process robustness, we investigated the possibility of replacing BSA and human transferrin present in the culture medium with purified recombinant products kindly provided by Novozymes Biopharma. In cultures containing recombinant human albumin and transferrin, we achieved a log-mean expansion of 2.60×108-fold compared to 5.63×108-fold for matched controls using BSA and donor-derived transferrin (n=3; p=0.51). No differences in enucleation characteristics were observed between the two medium formulations.
Discussion
While manufactured RBC have been suggested as an alternative to limited supplies of donor product, the quantities of cells required for therapeutic use are very large. Before pursuing such a strategy, two basic questions must be addressed: (1) Are there any fundamental barriers to large-scale manufacture of RBC? (2) Is it possible to generate clinically relevant numbers of cells?
With one exception, 6 existing methods of ex vivo RBC production employ static culture in plastic flasks. Due to mass transport limitations, static cultures typically only support cell densities up to a maximum 5×106/mL and a large air–medium interface is required for diffusion. To produce a single unit of RBC in standard tissue culture flasks would, at minimum, require a 400 L culture and ∼100 m2 of surface area. Clearly, this is not feasible.
The typical approach to cell culture scale-up is to move from static flasks to convectively mixed bioreactors. While in principle this is relatively straight forward to achieve, the use of feeder-layers is a substantial complication. Giarratana et al. 3 reported that eliminating direct contact between ex vivo generated erythroid progenitors and feeder-cells abolished enucleation and induced cell lysis, suggesting that direct interaction with stromal elements is essential for functional maturation of erythroid progenitors. This implies that for maximum efficiency, erythroid cells must be in physical contact with the feeder-layer, an arrangement that is difficult to achieve in large-scale bioreactor systems.
Although Miharada et al. 5 showed that feeder-layers are not strictly necessary, their cell yields were equivalent to only 1.75 units/UCB donation and enucleation was less efficient. Similarly, Leberbauer et al. 8 reported that only partial hemoglobinization was achieved (∼40% of donor levels) in feeder-free cultures, and the frequency of enucleation was not stated.
In contrast to the reports described above, we show that not only are feeder-layers dispensable, but that expansion and enucleation proceed efficiently in their absence. Over the same duration of culture, we achieved numerical expansions 1.73×106-fold in feeder-free culture (Fig. 1A) compared to the 1.95-fold×106-fold expansion reported by Giarratana and colleagues. 3 We observed an enucleation frequency of greater than 90% (Fig. 2B) and obtained a fully hemoglobinized cell product.
Having eliminated the use of feeder-layers, we undertook to translate our culture method for use in an agitated bioreactor system. The use of stirred tank bioreactors for hematopoietic stem cell cultivation was first reported in the early 90s,11,12 and shortly thereafter stirred cultivation in serum free conditions was described. 13 These and numerous subsequent articles have, however, typically focused on the generation of products for use in a stem cell transplantation setting, or immature progenitor cell products for the support of cytopenic postcancer-therapy or bone marrow transplant patients. While significant in these contexts, the degree of numerical expansion obtained was, however, insignificant relative that required for a mature RBC product.
A key aspect of maximizing the expansion of RBC in our protocol and others,5–7 is the initiation of cultures at very low cell densities (10,000 cells/mL). We have previously found that agitation of CD34+ cell cultures at these low densities is highly detrimental. 9 Consequently, we developed a two-stage culture protocol where the initial low density cultures are maintained under static conditions, and subsequently transferred to agitated culture once densities have increased to more typical levels. 9 When applying this same method for initial scale-up of RBC cultures, we observed that expansion was still dramatically reduced (33%) compared to static cultures. This did not appear to be a result of cellular damage/stress; however, it did appear that differentiation proceeded more rapidly in these cultures. Boehm et al. 6 also observed an apparent acceleration of differentiation in agitated cultures compared to static controls. They hypothesized that this may be due to the altered dynamics of cell–cell interactions and substrate adhesion in agitated culture. In contrast, we believed that this may be due to the higher DO levels experienced by cells in agitated culture compared to static culture. DO levels have been shown by others to alter the rate and outcome of hematopoietic differentiation in vitro.14–16 By reducing the DO content in our two-phase bioreactor process, expansion potential was restored and performance was similar to that for static controls (Fig. 3).
We next turned our attention to the issue of limited cell yield. Given that UCB transplants are able to support life-long hematopoiesis in recipients, it seemed probable that the limited ex vivo RBC yields achieved to date are a consequence of sub-optimal performance rather than biological capacity. In the feeder-dependent conditions of Giarrtana et al., 3 UCB-derived RBC cultures entered stationary phase before day 16. In contrast, Leberbauer et al. 8 achieved numerical expansions of 108–109-fold in feeder-free cultures over a 45-day period. In these prolonged cultures, however, the expanded cells did not fully mature and limited terminal differentiation required medium supplementation with 3% serum.
In line with the findings for Leberbauer et al., 8 data for our feeder-free method indicated that at day 21 cultures were still actively expanding (Fig. 1A). When we extended cultures through to day 33, a 2.25×108-fold expansion in cell number was achieved (Fig. 4). This is equivalent to more than 500 units of RBC per UCB donation. Although cultivation for longer durations was possible (in one case giving 1.68×1010-fold expansion at day 55), we observed that subsequent enucleation was inconsistent (and typically poor) and cells lacked the deep red coloring characteristic of shorter duration cultures. We speculate that these prolonged cultures in the presence of high cytokine concentrations may lead to transformation of the cells and loss of differentiation potential.
In addition to scale-up and cell yield, product quality and safety is an important consideration. To improve process robustness and eliminate the risk of disease transmission, fully defined culture medium that is free from animal- or human-derived products should be ideally be employed. Although serum-free, previously described erythroid culture media contain animal- or donor-derived albumin/plasma and transferrin. We investigated the possibility of directly substituting these components for commercially produced recombinant equivalents. These substituted cultures performed similarly to our standard formulation, with no further modification to either the medium or culture protocol required. In contrast, we have tested both human and bovine albumin from several different suppliers and observed large differences in performance (data not shown). This likely reflects different methods and extents of purification.
Conclusions
We have simultaneously demonstrated that (1) feeder cells are not required for efficient expansion and terminal differentiation; (2) process scale-up can be achieved in convective bioreactor systems; (3) clinically meaningful cell yields can be obtained from single UCB donations; and (4) this can be achieved in fully defined animal component free culture medium. Due to the large number of cells that are required, substantial challenges still exist with respect to process engineering and production costs.4,17 At this early stage, however, there appear to be no fundamental barriers to realizing industrial manufacture of RBC.
Footnotes
Acknowledgments
This work was supported by the Australian Stem Cell Centre (Stream 1, Module 4). We gratefully acknowledge the assistance of staff at the Royal Brisbane and Women's Hospital maternity suite, and the generosity of the donors.
Disclosure Statement
The authors declare no conflict of interest.