
Abstract
Achievements in tissue engineering using mesenchymal stem cells (MSC) demand a clinically acceptable “off-the-shelf” cell therapy product. Efficacy of cryopreservation of human bone marrow-derived MSC in clinically safe, animal product-free medium containing 2%, 5%, and 10% dimethyl sulfoxide (DMSO) was evaluated by measuring cell recovery, viability, apoptosis, proliferation rate, expression of a broad panel of MSC markers, and osteogenic differentiation. Rate-controlled freezing in CryoStor media was performed in a programmable cell freezer. About 95% of frozen cells were recovered as live cells after freezing in CryoStor solutions with 5% and 10% DMSO followed by storage in liquid nitrogen for 1 month. Cell recovery after 5 months storage was 72% and 80% for 5% and 10% DMSO, respectively. Measurements of caspase 3 activity demonstrated that 15.5% and 12.8% of cells after 1 month and 18.3% and 12.9% of cells after 5 months storage in 5% and 10% DMSO, respectively, were apoptotic. Proliferation of MSC recovered after cryopreservation was measured during 2 weeks post-plating. Proliferation rate was not compromised and was even enhanced. Cryopreservation did not alter expression of MSC markers. Quantitative analysis of alkaline phosphatase (ALP) activity, ALP surface expression and Ca++ deposition in previously cryopreserved MSC and then differentiated for 3 weeks in osteogenic medium demonstrated the same degree of osteogenic differentiation as in unfrozen parallel cultures. Cell viability and functional parameters were analyzed in MSC after short-term storage at 4°C in HypoThermosol-FRS solution, also free of animal products. Hypothermic storage for 2 and 4 days resulted in about 100% and 85% cell recovery, respectively, less than 10% of apoptotic cells, and normal proliferation, marker expression, and osteogenic potential. Overall, our results demonstrate that human MSC could be successfully cryopreserved for banking and clinical applications and delivered to the bedside in clinically safe protective reagents.
Introduction
Despite the progress in experimental regenerative studies and accumulating data on possible applications of MSC as immunomodulators and sources of trophic factors in various clinical conditions, 8 clinically accepted methods of cryopreservation and storage of MSC are poorly developed. Standardized procedures for clinical grade cryopreservation of MSC are needed for accumulation of large numbers of autologous cells for one time or repetitive transplantations during a long process of tissue repair, especially for bone regeneration, and also for creation of banks of allogeneic MSC.
A defined medium for a short storage of MSC without freezing is also of importance, as it will improve transportability of stem cells and will allow the necessary time for the completion of safety and quality control tests.9,10
Many researchers have previously reported that cryopreserved MSC maintain their potential for proliferation and osteogenic differentiation in vitro11–17 and in vivo.18,19 However, the non-approved reagents were used in most cases without procedural optimization and validations. 20 In a recent study sponsored by EU consortium “Crystal” (CRYo-banking of Stem cells for human Therapeutic AppLications), a cryopreservation procedure in an animal product-free and chemically defined cryopreservation media has been described for umbilical cord blood-derived erythroid and endothelial progenitors as well as adipose tissue mesenchymal stromal cells. Recovery and differentiation potential after cryopreservation were analyzed with promising results. 21
In the current study, cryopreservation of human BM-derived MSC in the commercial xeno-free media containing 2%, 5% and 10% dimethyl sulfoxide (DMSO) has been optimized and tested with emphasis on cell recovery, apoptosis, long-term proliferation kinetics, marker expression, and quantitative analysis of osteogenic differentiation. Rate-controlled freezing of MSC was performed using different regimens. The results of the study demonstrate good and reproducible physical and functional recovery of cryopreserved MSC after 5 months storage in liquid nitrogen, followed by optimized thawing procedure.
MSC were also subjected to a short-term storage at 4°C in a xeno-free HypoThermosol medium. Quantitative viability and functional tests were performed before and after storage in the same MSC cultures. We demonstrate that hypothermic storage of MSC in this animal product-free medium for 4 days does not affect MSC viability, proliferation, and osteogenic potential.
Materials and Methods
Isolation of MSC
BM aspirates were purchased from Lonza, USA, and delivered within 36 h to Israel at 4°C. MSC were isolated according to adhesion selection method as described elsewhere. 22 As previously shown, these cells expressed the main markers of MSC, were negative for CD45, and were capable of differentiation into osteoblasts, adipocytes, and chondrocytes. 22 Cells were passaged in MSC growth medium (GM) purchased from Lonza by plating 5000 cells/cm2. The average yield of MSC at confluency was 50–60,000 cells/cm2, suggesting 3–4 cell doublings between passages. Cells of passages 2–4 that had not been previously frozen were used in all the experiments.
Cryopreservation protocol
MSC were re-suspended in cold animal product-free cryopresrvation media: CryoStor-2 (CS-2), CryoStor-5 (CS-5), and CryoStor-10 (CS-10) containing 2%, 5%, and 10% DMSO, respectively (BioLife Solutions Inc.), or in conventional freezing medium (FM; 90% complete GM from Lonza containing 10% fetal calf serum [FCS]/30% by weight bovine serum albumin [BSA]/10% DMSO) at 106 cells/mL and aliquoted into cryo-vials (NUNC/Thermo Fisher Scientific) at 0.5 mL/vial (500,000 cells). The vials were pre-cooled on ice for 10 min and then placed in the programmable cell freezer Kryo-560-16-230 (Planer PLC). Samples were slowly cooled to −5°C (freezing point for CryoStor reagents) and then given a blast of chilling to −25°C and quick return to −5° C to prevent super-cooling and to ensure extracellular ice nucleation. From that point, samples were slowly cooled to −60°C at 1°C/min, and then, fast frozen at −25°C/min down to −196°C. A temperature sensor was inserted in a cryo-vial containing an appropriate cryo-medium. Frozen cells were stored in liquid nitrogen (liquid phase) for about 1 month or for 5 months.
Thawing procedure
Cells were thawed fast in a 37°C water bath with gentle agitation without allowing the sample to warm above chilled temperatures (0°C −10°C; until ice crystals still remained visible) and then gradually (drop wise) resuspended in cold GM in order to avoid drastic changes in osmolarity and prevent toxicity of cryo-protective reagents. Cell suspension in GM was centrifuged at 250 g and resuspended in 0.5 mL phosphate buffer saline (PBS) containing calcium and magnesium. The cell suspension in PBS was aliquoted as follows: a 100 microL suspension was taken for a Live/Dead assay (see below), and 300 microL were taken for apoptosis assay. The rest of the cells were used for plating for proliferation and differentiation assays at 3000 cells/cm2 based on live cell counts with Trypan blue.
Live/dead assay
Immediately after thawing, 100 microL of cell suspension in PBS was mixed in a 24-well plate with 0.5 mL PBS (with Ca++ and Mg++) containing 5 microM of Calcein-AM (Invitrogen), an indicator of live cells, and 10 microM of Ethidium homodimer-1 (Invitrogen), an indicator of dead cells. The plate was incubated in a CO2 incubator at 37°C for 35 min, then centrifuged at 250xg for 5 min to pellet the cells to the bottom in order to get more precise fluorescence readings. Cell fluorescence was measured on a Synergy-BioTek plate reader with a fluorescence detector at the bottom of the plates at an excitation/emission of 485/530 nm and 530/645 nm for Calcein-AM and Ethidium respectively. Background fluorescence of a PBS solution containing 5 microM Calcein-AM and 10 microM Ethidium was subtracted from each value. Live cell number detected by Calcein AM was calculated based on a standard curve (see Proliferation Assay next). The maximal Ethidium fluorescence signal was obtained by permeabilization of 100,000 MSC with 0.1% saponin. Percentage of dead cells was calculated as follows:
where Fx was fluorescence of a cell sample, Fbckgr was fluorescence of the background wells without cells, and Fmax was a fluorescence of the same number of cells (100,000) treated with 0.1% saponin.
Measuring cell proliferation with Calcein-AM
Thawed MSC were plated in duplicates into 24-well plates at 3000 cells/cm2 based on the Trypan blue counts of live cells. Cells were allowed to proliferate for 14 days in GM with medium changed twice a week. Cell number was measured the next day after plating (day 1), and on days 4, 7, and 14. At each time point, a number of viable cells was measured by Calcein-AM staining. For the standard curve, serial dilutions of unfrozen live MSC pre-stained with Calcein-AM were plated onto a 24-well plate. The cells were spun down to the bottom of the wells, and Calcein-AM fluorescence was measured.
Measurement of apoptotic cells: flow cytometry analysis of Annexin-V binding
The AnnexinV-FITC kit (IQProducts) was used. Thawed MSC suspension were immediately labeled with Annexin V according to manufacturer's instructions and analyzed on FACSAria (Becton Dickinson Biosciences) using FITC channel.
Measurement of apoptotic cells: flow cytometry analysis of Hoechst staining
Thawed MSC were resuspended in fluorescence-activated cell sorter (FACS) buffer containing 10 microg/mL Hoechst 33342 (Sigma) at 200,000 cells/200 microL. Cell suspension was incubated for 10 min at 37°C in the dark and then analyzed on FACSAria.
Measurement of apoptotic cells: flow cytometry analysis of caspase 3 activity
The EnzChek Caspase 3 Assay Kit (Invitrogen) for adherent cells was modified for flow cytometry. Cryopreserved 300,000 MSC were thawed and immediately resuspended in 300 microL Pipes/EDTA/Chaps reaction buffer supplemented with 3 microL dithiothreitol (no previous lysis of cells was performed); then, 1.5 microL Z-DEVD-R110 rhodamine substrate was added to cell suspensions. The samples were incubated for 20 min at 37°C in the dark, and transferred on ice to stop the reaction. Two controls were performed: (1) cells plus substrate reaction mixture were kept on ice for 20 min and (2) DMSO was added instead of a substrate, and samples were incubated for 20 min at 37°C. In some experiments, cells were preincubated with 1.5 microL of the 1 mM stock solution of the caspase 3 specific inhibitor Ac-DEVD-CHO for 10 min at 37°C and then incubated with caspase 3 rhodamine substrate. At the end of the incubation, 7-AAD (eBioscience) was added to the reaction mixture (15 microL of the 50 microg/mL stock solution per sample) for detection of dead cells. The samples were analyzed on FACSAria using the FITC channel for measuring fluorescence of the caspase 3 product and PerCP channel for 7-AAD.
Hypothermic storage protocol
MSC cultures were grown to 70%–80% confluency, and GM was replaced with pre-cooled HypoThermosol-FRS medium (HTS-FRS, BioLife Solutions). Cell cultures were stored at 4°C for 2 and 4 days in the laboratory refrigerator with a temperature control and alarm system. At the end of the storage period, HTS-FRS medium was replaced with warm GM, and the cultures were moved to a CO2 incubator and allowed to recover at 37°C for 3 h. At the end of the recovery period, the cells were detached by trypsinization and used for flow cytometry analysis of dead cells (7-AAD staining), apoptotic cells (caspase 3 activity), and expression of MSC markers. For proliferation and differentiation assays, MSC were subjected to hypothermic storage and recovery and then continued growing in GM and osteogenic medium (OM), respectively, without replating.
Measuring live cells with Alamar Blue
In order to measure the number of live cells before and after hypothermic storage in the same cultures, an oxidation-reduction indicator Alamar Blue (AbD Serotec) that chemically interacts with metabolic products released by living cells into the culture medium was used. MSC were plated in duplicates into 24-well plates at 3000 cells/cm2 and allowed to grow to subconfluency. Right before hypothermic storage, a 0.5 mL/well of fresh GM containing 10% Alamar Blue was added to the cells, and plates were placed for 2 h in a cell incubator. At the end of the incubation, the medium containing Alamar Blue was transferred into a new 24-well plate, and its fluorescence was measured on a Synergy-BioTek plate reader at an excitation/emission of 530/590 nm. HTS-FRS was added to the cells, and the cells were stored for indicated times at 4°C. At the end of the storage and recovery, the number of live cells was measured again with Alamar Blue. A standard curve was produced by plating MSC in serial dilutions into 24-well plates. Cells were allowed to adhere overnight in the cell incubator. The next day, a fresh medium with 10% Alamar Blue was added to the cells, and Alamar Blue fluorescence was measured. In order to account for possible cell proliferation during the overnight adhesion period, the live cell number was measured with Calcein-AM and plotted against Alamar Blue fluorescence. The standard curve was highly linear (R2=0.9952).
Staining cells with antibodies against MSC markers
Immediately after thawing, MSC were resuspended at 106/mL in FACS buffer [2% BSA (Sigma), 2% human blocking serum (Chemicon) in DMEM without phenol red (Sigma)], and aliquoted at 100 microL per well (100,000 cells) onto a polypropylene U-shaped 96-well plate. Each cell aliquot was incubated with saturating concentrations of antibodies against MSC markers or isotype-matched controls on ice for 30 min; then, the plate was centrifuged at 200xg for 3 min and inverted onto a paper towel to drain the supernatant. The cells were washed twice in 200 microL FACS buffer and transferred into a polypropylene FACS test tube containing 500 microL FACS buffer. Analysis of the results was performed using DIVA software (Becton Dickinson). The list of antibodies used in the experiments is shown in Table 1.
APC, antigen presenting cells.
Osteogenic differentiation of MSC
Thawed MSC were plated in GM overnight, and then the GM was replaced either by OM (alpha-MEM [Sigma], 10% FCS heat inactivated [Biological Industries], 0.2 mM L-ascorbic acid-2-phosphate [Mg salt n-hydrated; Fluka], 10 mM Glycerol 2-phosphate disodium salt hydrate [Sigma], 100 nM dexamethasone [Sigma], 2 mM GlutaMAX™-I Supplement [Invitrogen], 100 units/mL penicillin, 0.1 mg/mL streptomycin, 0.25 mg/mL amphotericin B) or by fresh GM for comparison. The cells were grown for various periods of time while the OM was changed twice a week.
Alkaline phosphatase activity assay
MSC cells growing in 24-well plates in OM and control cultures growing in GM were lysed on days 7 and 14 with 500 microL cold lysis buffer (1 mM MgCl2/0.5% Triton×100 in Alkaline Buffer Solution [Sigma]) and incubated on ice for 1 hour. The lysates were centrifuged at 13,000 rpm for 2 min, and 100 microL cell lysates were combined with 400 microL Phosphatase Substrate Solution (20 mg/mL of p-nitrophenol phosphate [p-NP; Sigma] in 5 mL Alkaline Buffer Solution diluted 1:3 in ddH2O) and incubated at 37°C for 10 min. The reaction was stopped with 500 microL EDTA-NaOH stop solution (20 g NaOH plus 37.22 g Na2EDTA in 500 mL ddH2O). 200 microL of each sample was transferred to a 96-well plate, and absorbance was read at 404 nm using a Synergy plate reader. The results were expressed as nmol p-NP/mL/min and normalized to the number of living cells in the corresponding wells determined by Calcein-AM assay as just described.
Alkaline phosphatase surface expression
MSC cells growing in 25 cm2 flasks in OM and control cultures growing in GM were trypsinized on day 14 and stained with antibodies against human alkaline phosphatase (ALP; liver/bone/kidney isozyme; see antibody information in Table 1). Stained cells were analyzed by flow cytometry.
Calcium deposition assay
MSC cells growing in 24-well plates in OM for 21 days and parallel cultures growing in GM were washed with PBS without Ca++, Mg++ and then lysed with 250 microL/well 0.5N HCl. The lysates were shaken at 4°C overnight to extract calcium and then centrifuged at 13,000 rpm for 2 min at 4°C. The assay was set up in 96-well plates using Calcium Liquicolor kit (Stanbio Labs) according to the manufacturer's instructions. The amount of Ca++ was normalized to the number of living cells in the corresponding wells determined by Calcein-AM assay as just described.
Statistical analysis
A comparison between control and experimental groups in parallel cultures was performed with Student's t-test. For multiple measurements, the Anova test was used.
Results
MSC injury induced by the slow freezing phase of the cryopreservation program
The graded freezing technique 23 was used to calibrate Ethidium assay and to differentiate between slow and fast freezing injury. MSC resuspended in various freezing solutions were frozen at −1°C/min to −60°C. At that temperature, the cells were immediately thawed and stained with Ethidium homodimer as described in Methods. In order to exclude possible toxic effects of cryo-solutions on cells during pre-cooling, parallel cultures were kept at 4°C. Maximal Ethidium fluorescence of the same number of cells permeabilized with 0.1% saponin was around 1200 relative fluorescence units (RFU; see Methods for details). As shown in Figure 1A, Ethidium fluorescence of cells frozen in GM without cryo-protectants was about 400 RFU, suggesting that about 30% of cells frozen without cryo-protectants had already died during the slow freezing phase. Fluorescence of MSC slowly frozen in conventional FM and in CryoStor reagents was not different from that of live cells kept in suspension in GM at 37°C during the duration of the slow freezing phase (see Fig. 1A). Keeping cells in suspension allowed for accounting for a possible cell death that occurs after MSC are kept in a non-adherent state for a prolonged time (our unpublished observations).
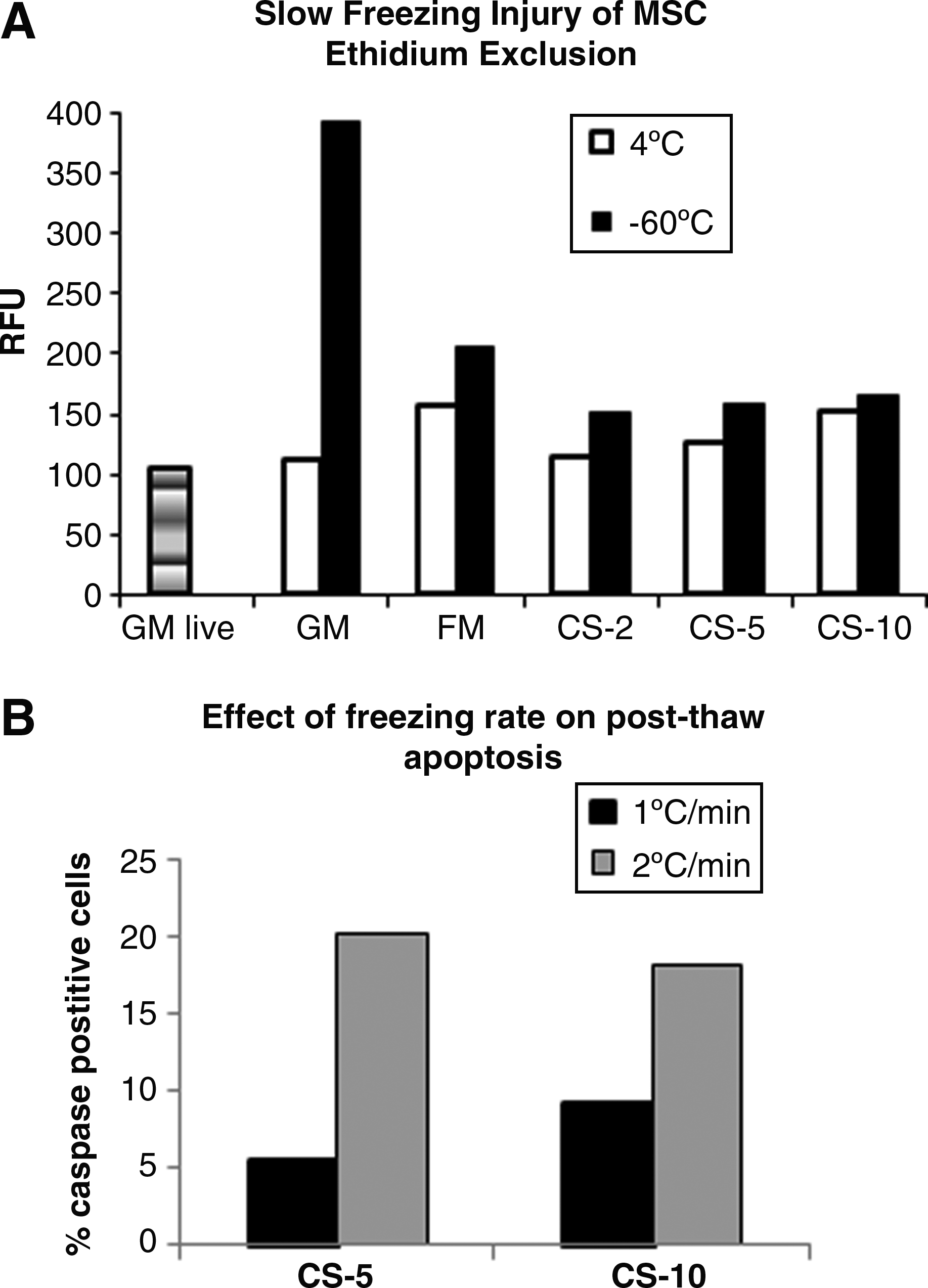
The effect of slow freezing rate on MSC viability.
Manufacturer instructions for CryoStor solutions suggested that after fast thawing, the cells could be diluted with GM in a single-step procedure. We have tested this protocol for MSC and compared it with an alternative procedure where thawed cells were diluted with GM gradually to minimize the osmotic injury. MSC were frozen to −60°C in the conventional FM and in CS-10, then quickly thawed at 37°C, and resuspended in 10 mL GM by adding it in either one step or stepwise. Ethidium fluorescence of the cells resuspended in GM in one step was about 800 RFU, while fluorescence of cells resuspended stepwise was similar to the live cell fluorescence (around 100 RFU), suggesting that stepwise addition of medium during MSC thawing is critical for minimizing osmotic injury.
Viability of MSC after cryopreservation for 1 and 5 months
Recovery of MSC after cryopreservation for 1 month in a CS-2 containing 2% DMSO was about 91.7%, while recovery of MSC cryopreserved in CryoStor solutions with 5% and 10% DMSO was above 95% (Table 2). Since CS-2 had inferior cryoprotective ability, only CS-5 and CryoStore-10 were tested during a 5 month cryopreservation. Percentage of viable cells after 5 month storage was lower than that of cells frozen for 1 month: 72% and 80%, respectively.
Mesenchymal stem cells (MSC) were frozen in growth medium (GM) without cryoprtectants, in a conventional freezing medium (FM), or in CryoStor-2 (CS-2), CryoStor-5 (CS-5), and CryoStor-10 (CS-10) reagents for 1 and 5 months. Percentages of live and dead cells were determined immediately after thawing before cell plating. Parallel cultures were analyzed before freezing.
Mean±SD of 4 experiments performed with 4 MSC batches each derived from a different donor. SD, standard deviation.
Mean±SD of 3 experiments performed with 3 MSC batches each derived from a different donor.
ND, not done.
Percentage of dead cells in samples cryopreserved in CryoStor reagents for 1 month ranged between 13% and 15% and was comparable to that of MSC frozen in conventional FM (17%), while samples frozen without cryo-protectants had significantly more dead cells (72%). (Table 2). Similar results were obtained for MSC cryopreserved for 5 months (Table 2).
MSC apoptosis after cryopreservation for 1 and 5 months
Calibration of caspase 3 assay for flow cytometry was performed using a sample of apoptotic MSC treated in a suspension with staurosporine, a PK-C inhibitor, and a pro-apoptotic agonist. 24 Representative histograms obtained for staurosporine-treated and control untreated cells are shown in Figure 2A. A top histogram was produced with freshly trypsinized MSC. The main peak gated on this histogram was defined as a background caspase 3 activity peak. All the events to the right of this gate were defined as cells with high caspase activity or apoptotic cells. All the events to the left of the background peak are mostly debris lacking caspase activity. The same gate was copied onto histogram produced with MSC incubated with 1 microM staurosporine for 4 h (Fig. 2A middle histogram). A sharp increase in the number of the events to the right of the gate (apoptotic cells with high caspase activity) was registered. When cells incubated with staurosporine were pretreated with caspase 3–specific inhibitor, the number of events with high caspase activity significantly decreased (Fig. 2A bottom histogram). A statistical analysis of the histograms is summarized in Figure 2B.
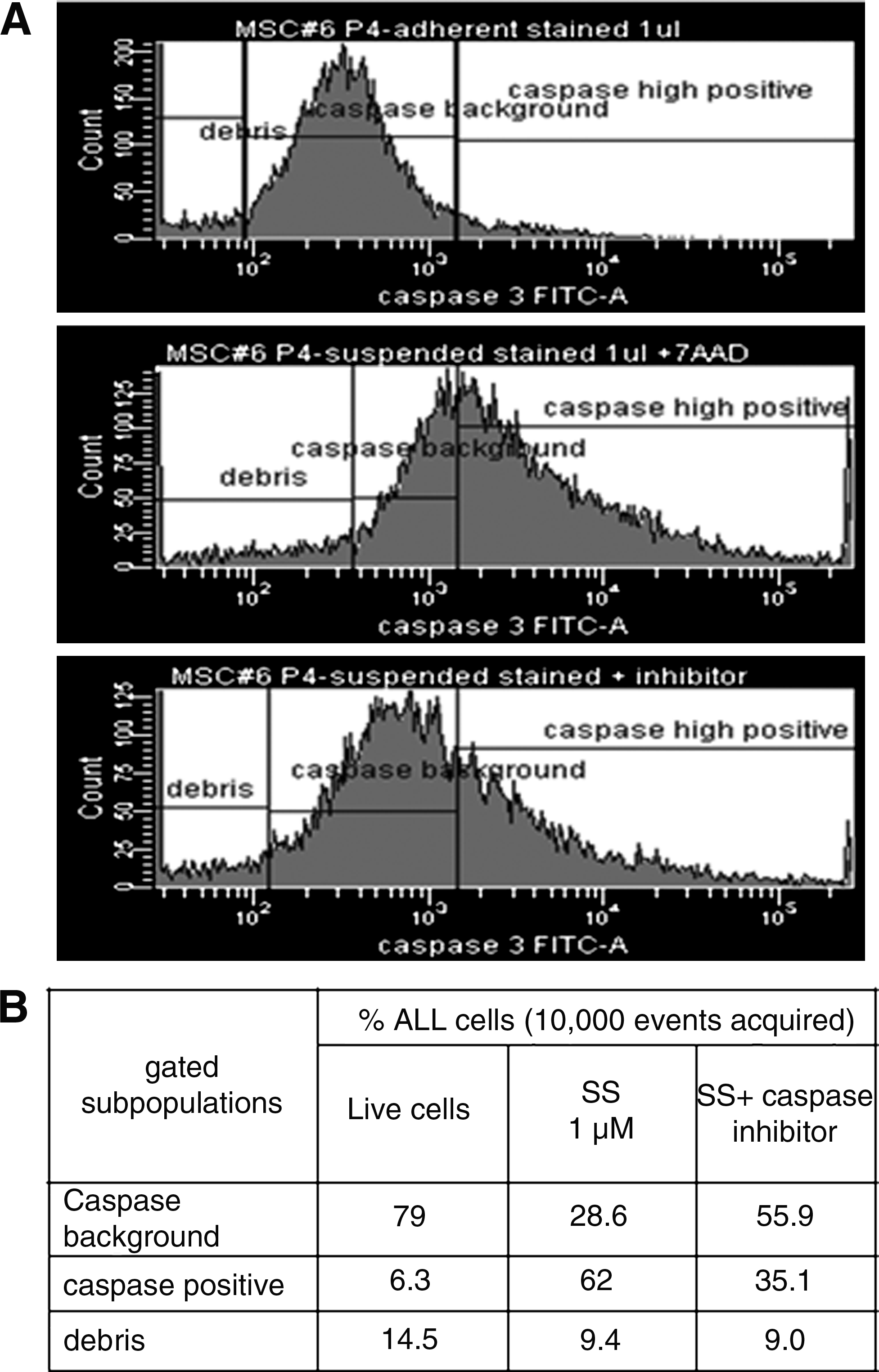
Calibration of the flow cytometry analysis of caspase 3 activity, a representative experiment.
The results of caspase 3 assay and also of Annexin V and Hoechst 33342 staining of apoptotic cells in MSC suspensions recovered after 1 month of storage in liquid nitrogen are summarized in Table 3. When apoptosis was evaluated by Annexin V or Hoechst 33342 staining, lower percentages of apoptotic cells were observed for all tested media, compared with the results of caspase 3 assay, and no significant differences between FM and CryoStor regents were found. However, when apoptosis was measured by caspase 3 assay, there was on an average 13%–17% apoptotic cells in samples previously cryopreserved in CryoStor reagents, while cryopreservation in conventional FM resulted in only 3% of apoptotic cells (Table 3). The differences between samples stored in CS-2, CS-5 and CS-10 were not statistically significant, while the differences between CryoStor reagents and conventional FM were statistically significant.
MSC were frozen in a conventional FM, or in CS-2, CS-5, and CS-10 reagents for 1 and 5 months. Percentage of apoptotic cells was determined immediately after thawing before cell plating.
For cells cryopreserved for 1 months, apoptosis was measured by 3 different methods; for cells stored for 5 months, apoptosis was measured only by caspase 3 assay, which had previously demonstrated the highest percentage of apoptotic cells.
When MSC were thawed after being cryopreserved in CS-5 and CS-10 for 5 months, similar levels of apoptotic cells compared with one-month storage were observed (Table 3).
Proliferation rate of MSC after cryopreservation for 1 and 5 months
Based on viability data showing a slightly lower viability of MSC cryopreserved with 2% DMSO, only MSC cryopreserved in CS-5 and CS-10 were used for assessment of cell proliferation. As shown in Figure 3, cryopreservation of MSC for both 1 and 5 months did not compromise and even enhanced cell proliferation. After expansion for 14 days in GM, the number of control unfrozen cells increased 13-fold, while the number of cells cryopreserved with CS-5 and CS-10 for 1 month increased 22 and 26.5-fold, respectively (Fig. 3A). Increased proliferation of MSC after cryopreservation was statistically significant and most probably occurred because of selection of “stronger” cells at plating. It is worth noticing that next day after plating (day 1) less cryopreserved than unfrozen MSC adhered to the plates: 5528±647 and 6417±2692 for MSC cryopreserved in CS-5 and CS-10, respectively, compared with 8789±2336 unfrozen cells (mean±SD; n=5). While the difference in seeded cell number between unfrozen cells and cryopreserved in CS-10 cells was not statistically significant, the difference between unfrozen cryopreserved in CS-5 cells was statistically significant (p≤0.02).

MSC proliferation of MSC cryopreserved for1 and 5 months. MSC were cryopreserved in CryoStor solutions (CS-5 and CS-10) and stored for 1 month
Similar results were obtained after cryopreservation for 5 months. Again, proliferation of cryopreserved cells was higher than that of unfrozen cells (Fig. 3B). Similarly, less adherent cells were found the next day after plating of MSC cryopreserved in CS-5 (3573±1622) and CryoStor10 (4277±1387) compared with unfrozen cells (6398±21; mean±SD; n=3). These differences in seeded cell numbers were compensated already by day 7 of culturing due to the higher proliferation rates of previously cryopreserved cells.
Osteogenic differentiation of MSC after cryopreservation for 1 and 5 months
MSC cryopreserved either for 1 month or for 5 months retained their osteogenic potential. As summarized in Table 4, all tested samples of MSC upregulated ALP activity 5-10-fold over undifferentiated cells cultured in parallel in GM. No significant difference between ALP activity of MSC cryopreserved in CryoStor reagents and unfrozen MSC was observed after both 1 and 5 months of storage in liquid nitrogen. CryoStor regents were equal to conventional FM, containing FCS in terms of osteogenic upregulation of ALP activity (see Table 4, 1 month cryopreservation).
MSC were frozen in a conventional FM, and in CS-2, CS-5, and CS-10 reagents for 1 and 5 months. After thawing, MSC were seeded in 24-well plates and incubated in osteogenic medium for 7 and 14 days. The data is ALP activity in nmol pNP/min/10,000 cells.
Mean±SD of 6 experiments performed with 6 MSC batches, each derived from a different donor. ALP activity in parallel MSC cultures kept in GM was 0.22±0.19 and 0.52±0.27 for 7 and 14 days, respectively.
Mean±SD of 3 experiments performed with 3 MSC batches, each derived from a different donor. ALP activity in parallel MSC cultures kept in GM was 0.32±0.24 and 1.88±0.91 for 7 and 14 days respectively.
Flow cytometry analysis of ALP surface expression measured on day 14 after addition of OM demonstrated that there was no difference in the percentage of ALP-positive cells between unfrozen control cells and those stored in CS-5 and CS-10 for one month (89.6%, 87.8%, and 97.8% respectively). Similarly, storage for 5 months did not affect upregulation of ALP expression: 78.1% of unfrozen MSC and 65.2% of MSC cryopreserved in CS-10 became ALP-positive after osteogenic stimulation for 14 days.
Ca++ deposition into extracellular space was measured after 21 days of osteogenic differentiation. No statistically significant differences in the extracted Ca++ concentrations (microM/10000 cells) between unfrozen cells (1.9±0.4) and cells cryopreserved for 1 month in CS-5 (2.0±0.7) or CS-10 (2.1±0.7; Mean±SD; n=3) were observed. Similarly, cryopreservation in CryoStor reagents for 5 months did not significantly affect Ca++ deposition, which was 1.0±0.2, 0.7±0.3 and 0.8±0.2 microM/10000 cells for unfrozen cells, and cells cryopreserved in CS-5 or CS-10, respectively (Mean±SD; n=3).
Analysis of expression of MCS markers after cryopreservation for 5 months
Flow cytometry analysis of MCS markers was performed after a 5 month storage in CS-10 and compared with marker expression in the same cells before freezing. Unfrozen MSC expressed high levels of all markers tested, except CD9 (the latter was expressed on 34% of MSC). This pattern of expression did not change after cryopreservation, except the percentage of CD9-positive cells dropped to 18% (Table 5).
All data are a percentage of cells positive for the corresponding marker; Mean±SD of 3 experiments performed with 3 MSC batches, each derived from a different donor.
MSC were frozen in CS-10 for 5 months. After thawing, MSC were seeded into cell culture flasks at 3000 cells/cm2, allowed to proliferate in GM for 1 week, and then stained with antibodies against MSC markers. Parallel cultures were stained before freezing.
Subconfluent cultures of MSC were stored in HypoThermosol-FRS medium (HTS-FRS) at 4°C for 4 days, then, the HTS-FRS medium was replaced with warm GM, the cultures were allowed to recover in a CO2 incubator for 3 h, trypsinized, and stained with antibodies against MSC markers. Parallel cultures kept in GM at 37°C all the time were analyzed in the same way.
Hypothermic storage
The same battery of tests (viability, proliferation, osteogenic differentiation, and marker analysis) was performed for evaluation of efficacy of HypoThermosol®-FRS (HTS-FRS) medium for a short-term storage of MSC at 4°C.
The number of viable cells was measured with Alamar Blue test in the same cultures before hypothermic storage (40004±8380) and after incubation at 4°C in HTS medium for 2 days (47638±9899) and 4 days (34092±7883). In order to evaluate proliferation ability of MSC after hypothermic storage, control cells and those stored in HTS medium for 2 and 4 days were further expanded in GM for 14 days without re-plating. The number of cells in parallel cultures that remained in a cell incubator all the time increased during 14 days about 1.8-fold (from 43482±6840 to 78106±4500); the number of cells that were first stored at 4°C in HTS medium for 2 days also increased 1.8-fold (from 47638±9899 to 85009±1658) and the number of cells that were first stored at 4°C in HTS medium for 4 days increased 1.9-fold (from 34092±7883 to 66402±4953; n=4).
Dead cells and apoptotic cells were measured at the end of the recovery period using 7-AAD and caspase 3 staining, respectively. An increase in the percentage of dead cells from 2.3% in control cultures to 16.8% in stored cultures (p=0.043) and apoptotic cells from 2.9% in control cultures to 6–8% in stored cultures (p=0.008) was observed (Fig. 4B).

Hypothermic storage of MSC. Subconfluent cultures of MSC were stored in HTS-FRS at 4°C for 2 and 4 days. At the end of the storage period, HTS-FRS medium was replaced with GM, and the cultures were moved to a CO2 incubator and allowed to recover at 37°C for 3 h. Control parallel cultures that were not subjected to hypothermic storage but were kept in GM in the cell incubator were analyzed in the same way.
In order to evaluate the osteogenic differentiation of MSC after hypothermic storage, control cells and those stored in HTS medium for 2 and 4 days were allowed to differentiate in OM for 14 days without re-plating. The results are summarized in Figure 4C.
No significant differences in ALP activity between control cells (5.6±1.7 nmol pNP/min/10,000 cells) and those stored in HTS for 2 and 4 days (6.2±1.6 and 6.5±1.4 respectively) were found. ALP activity in parallel cultures of both control and stored cells that were not differentiated but kept for 2 weeks in GM was very low (Fig. 4C).
Similarly, ALP surface expression in control and stored cultures was not different. In control cultures, 70.2%±18.3% of differentiated cells were ALP positive; in cultures stored in HTS for 4 days and then differentiated, 61.6%±25.9% cells were ALP positive (mean±SD; n=3).
Finally, the expression of MSC markers was analyzed in MSC at the end of the recovery period. No differences between control and stored cells were found except the expression of CD9 decreased (see Table 5).
Discussion
To our knowledge, this is the first study examining in depth the efficacy of long-term cryopreservation of human BM-derived MSC in an animal product-free clinically accepted medium. Commercial CryoStor™ cryo-medium developed by BioLife Solutions that was chosen for this study has been in the market for some time and is distributed by several vendors, including key companies specializing in stem cell products. However, the efficacy of this medium for cryopreservation of human MSC has been poorly investigated, and no long-term cryopreservation studies were performed. In a recent study under the aegis of European consortium “Crystal” (CRYo-banking of Stem cells for human Therapeutic AppLications), xeno-free chemically defined media containing Pluronic F-68 and variable percentages of DMSO (0%, 5% and 10%) were tested for cryopreservation of various progenitor cells, including adipose tissue-derived mesenchymal stromal cells. A CryoStor solution containing 10% DMSO was also considered for this study; however, it was ruled out after pilot experiments. 21
The goal of the study was three-fold: (1) to develop and standardize a xeno-free cryopreservation protocol customized for MSC; (2) to optimize a battery of quantitative tests for evaluation of MSC viability, proliferation, and differentiation in order to evaluate the cryo-protocol efficacy; and (3) to assess the efficacy of MSC xeno-free cryopreservation after prolonged (5 months) storage in liquid nitrogen using these tests.
In the preliminary experiments conducted to optimize the freezing/thawing protocol and evaluation parameters, MSC were stored in liquid nitrogen for one month. In most of these experiments, the efficacy of CryoStor reagents was compared with the cryoprotective effect of the commonly used MSC FM formulated by Lonza on the basis of their MSC GM with addition of 10% FCS, 30% BSA, and 10% DMSO. Since this medium has been successfully tested in multiple labs including ours and in many research applications, it was chosen as a gold standard, although it cannot be used in clinical studies because of the presence of animal proteins. CryoStor reagents formulated without FCS, and other animal products were practically equal to FM, except that the number of apoptotic cells measured by caspase 3 activity was significantly lower in post-thaw cells cryopreserved in FM than in MSC cryopreserved in CryoStor reagents (see Table 3). However, one cannot exclude that this difference was due to the faster death of the apoptotic cells during cryo-storage in FM. When apoptosis was evaluated by Annexin V binding or by staining with Hoechst 33342, lower percentages of apoptotic cells were observed for all the media, and no significant differences between FM and CryoStor regents were found. One possible explanation for these results is that translocation of phosphoserine to the outer layer of the cell membrane detected by AnnexinV and slowing of Hoechst efflux resulting in its accumulation in apoptotic cells occurs at early stages of the apoptotic program 25 and then reverse or become shielded by further cell deterioration, while high Caspase 3 activity is detected at the later stage of apoptosis.
Evaluation of dead cells in the MSC samples thawed immediately after being slowly frozen to −60°C allowed isolation of cell injury, which occurs during slow cooling phase of the cryoprotocol. These experiments demonstrated the importance of MSC protection during slow cooling, and confirmed the high sensitivity of MSC to the rate of freezing: the increase of the freezing rate only from 1°C/min to 2°C/min caused a drastic increase in cell apoptosis (see Fig. 1B). It also suggests that the apoptotic program in MSC already starts during the slow cooling phase of cryopreservation and supports the findings of Carvalho et al., who demonstrated the progressive loss of MSC viability at cooling rates of −3°C/min, −5°C/min, and −10°C/min. 26
Another sensitive step of cryopreservation protocol is the thawing procedure. Contrary to the manufacturer's instructions, slow cooling injury of MSC was further exacerbated by osmotic injury resulting from fast addition of the dilution medium (GM) to the thawed cells, suggesting high sensitivity of MSC to changes in osmotic pressure.
A successful cryopreservation program commonly focuses on cell recovery scores. However, a significant number of recovered cells are often not able to adhere to the cell culture surface and can be found floating the next day after plating. Considering a well-established contribution of apoptosis to cryoinjury, 27 one cannot exclude that at least a part of the so-called “floating cells” are those cells in which cryopreservation triggered an apoptotic program. Revealed by caspase 3 assay, a relatively low percent (13%–17%) of apoptotic cells in post-thaw suspensions of MSC is consistent with the presence of anti-apoptotic agents in CryoStor solutions. 28 However, these apoptotic cells will most probably die after plating and, thus, should be taken into account when estimating the numbers of cryopreserved MSC for transplantation.
Evaluation of cell recovery, dead cell, and apoptotic cell number performed after complete cryopreservation program (slow and then fast cooling, followed by storage of the cells in liquid nitrogen and then gentle thawing) demonstrated a practically full recovery and high viability of MSC stored in liquid nitrogen for one month and about 70%–80% recovery after 5 month storage (see Table 2). One could notice that percentages of dead cells shown in Table 2 were higher than one would expect based on percentages of live cells, which was most probably due to the loss of a portion of cells in the sample permeabilized with 0.1% saponin, which was considered a 100% dead cell sample and which produced maximal fluorescence signal used for calculation of percentage of dead cells (see Methods). This assumption is supported by a comparable percentage of dead cells in control unfrozen samples (14%).
Besides testing viability, proliferation of MSC cells post cryopreservation was analyzed. The goal was to estimate proliferation rate over an extended time after plating rather than recovery of the cell division function within a few days after thawing as was done in the study just cited. 21 We show here for the first time that proliferation rate of post-cryopreserved MSC (after both 1 month and 5 month storage) is significantly higher than that of unfrozen control cells (Fig. 3A, B), suggesting possible cell selection. The higher proliferation rate was observed despite the fact that the next day after plating, less cryopreserved cells compared with unfrozen cells adhered to the culture plates, further supporting the hypothesis of selection of “stronger” cells after cryopreservation. This means that in the future clinical studies, when MSC transplantation is planned immediately after thawing, QC tests of cell viability and apoptosis should be performed before cell plating.
In addition, it is worth noting that comparison of expression of MSC markers in unfrozen cells and cells after cryopreservation (and after hypothermic storage) demonstrated no changes except in expression of CD9, which was downregulated post-freezing (see Table 5). We have previously shown that expression of CD9 in MSC and in fibroblasts has inverse relationship where MSC express low levels of CD9 and fibroblasts express high levels. 22 Furthermore, it was shown that in the higher passages of MSC (p6), CD9 expression goes up (from 39.4% to 73.5% CD9 positive cells, ref. 21). In the current study, we used MSC of passage 2–3 where CD9 expression was low. A decrease in the percentage of CD9-positive cells observed after cryopreservation or storage further supports selection of the “younger” phenotype. However, the exact changes occurring in more proliferative post-cryopreserved MSC remain obscure and should be addressed in the future most probably at the gene level. Tumorigenicity of post-cryopreserved MSC should also be challenged.
Since this lab has been focusing on osteogenic differentiation of MSC, we had at hand a number of quantitative assays for a thorough analysis of osteogenic differentiation, developed, optimized, and standardized over the years. These methods allowed us to reveal even subtle changes in cryopreserved cells, which would not have been possible to demonstrate by simple cytochemistry staining commonly used for demonstration of three lineage differentiation of MSC, especially considering variability of MSC batches. All quantitative tests (ALP activity, ALP surface expression, and mineralization of extracellular space) demonstrated no differences in osteogenic potential of cryopreserved MSC (whether stored in liquid nitrogen for 1 or for 5 months) and their paired unfrozen controls. Our findings are consistent with those of Woods and colleagues, who used CryoStor solution with 10% DMSO to preserve dental pulp and then demonstrated good three lineage differentiations of stem cells derived from previously cryopreserved dental pulp. 13 Regeneration of non-union bone fractures is a long process taking months and in cases of substantial bone loss, even years. It is likely that repetitive MSC transplantations will be necessary to accomplish full healing. Therefore, successful clinical grade cryopreservation of MSC for bone repair is the most urgent need. We hope that our multifactorial analysis of CryoStor cryo-medium for bone engineering will encourage a similar evaluation of this medium for application of MSC in various other fields of tissue engineering.
Post-cryopreservation viability, apoptosis, proliferation, marker expression, and differentiation of human stem cells from apical papilla have been recently studied by Ding et al., who demonstrated successful cryopreservation of these MSC using DMSO, ethylene glycol, and glycerol as cryoprotectants; however, FM in these studies contained 90% FCS. 29 Our findings clearly demonstrate that all these parameters are relatively well preserved in MSC cryopreserved in a clinical grade medium without serum and other animal products.
In a separate set of experiments, hypothermic storage of MSC in HypoThermosol-FRS™ storage/shipment medium also developed by BioLife Solutions was investigated. HypoThermosol medium was tested for hypothermic preservation of multiple cell types such as cardiac myocytes, 30 coronary vessels, 31 human renal cells, 32 and canine kidney cells 33 ; however, storage of MSC in HTS has not been previously studied. We have tested MSC storage in HTS for 2 and 4 days at 4°C. The same battery of tests was applied.
In order to test cell recovery in the same culture, live cell number was measured before and after storage with Alamar Blue indicator. The number of viable cells did not change significantly after 2 days of hypothermic storage and slightly decreased after 4 days of storage. Hypothermic storage of MSC in HTS for 6 days resulted in a significant decrease of cell viability (data not shown). It is worth noticing that MSC stored at 4°C in a regular GM died very quickly (data not shown). HTS was shown to preserve MSC marker expression, proliferation and osteogenic differentiation after storage for at least 4 days.
ALP activity of MSC after storage in continuous cultures in GM was quite low (see Fig. 3C), suggesting that HTS storage did not promote spontaneous differentiation of MSC.
An attempt was previously made to store porcine hepatocytes at 4°C in various preservation solutions, but none of the tested reagents was capable of adequately preventing cell death after 1 day of hypothermic storage and subsequent 1 day culturing in normal conditions. 34 In another report, human tubular epithelial cells stored for 2 days in the University of Wisconsin solution (UWS) followed by 1 day re-warming in GM showed increase of apoptotic signaling that was suppressed by addition of antioxidants to storage solution. 35 When UWS storage solution was compared with HTS-FRS, cell viability, energy status, and xenobiotic metabolism were consistently higher and, in some cases, markedly higher after storage in HTS-FRS. 36 In this study in addition to good recovery of MSC after hypothermic storage in HTS, percentage of apoptosis in stored cultures was also low. This could be explained by the presence of anti-oxidants and anti-apoptotic additives in HTS-FRS. 37 In summary, our results indicate that MSCs could be stored at 4°C for at least 4 days in a solution free of animal products.
In conclusion, we have critically evaluated clinical grade media for cryopreservation and short-term storage of human MSC. We have used a programmable cell freezer and an optimized freezing and thawing program. Quantitative tests of cell viability and apoptosis were adapted for MSC still remaining in a post-thaw suspension. Enhanced proliferation rates of post-cryopreserved MSC were observed and should alarm a scientific community. We believe that this study will contribute to acceleration of MSC application for treatment of non-union bone fractures and other types of tissue injury. Successful cryopreservation and storage of MSC should promote the creation of MSC banks. A standardized protocol for MSC cryopreservation will also resolve the variability of the data between different laboratories and enhance collaborations within the research community.
Footnotes
Acknowledgments
This work was supported by The Chief Scientist Office of the Israeli Ministry of Industry, Trade and Labor and The Israeli Consortium “Bereshith” for Cell Therapy
The experimental work presented in this article was performed while all the authors were working at Teva Pharmaceutical Industries, Petach Tikva, Israel. The authors want to thank Drs. Aharon Schwartz, Shoshana Merchav, and Doron Shinar of Teva Pharmaceutical Industries for their support toward this study.
Disclosure Statement
No competing financial interests exist.