
Abstract
A general problem in tissue engineering is the poor and insufficient blood supply to guarantee tissue cell survival as well as physiological tissue function. To address this limitation, we have developed an in vitro vascularization model in which a decellularized porcine small bowl segment, representing a capillary network within a collagen matrix (biological vascularized scaffold [BioVaSc]), is reseeded with microvascular endothelial cells (mvECs). However, since the supply of mvECs is limited, in general, and as these cells rapidly dedifferentiate, we have applied a novel technology, which allows the generation of large batches of quasi-primary cells with the ability to proliferate, whilst maintaining their differentiated functionality. These so called upcyte mvECs grew for an additional 15 population doublings (PDs) compared to primary cells. Upcyte mvECs retained endothelial characteristics, such as von Willebrandt Factor (vWF), CD31 and endothelial nitric oxide synthase (eNOS) expression, as well as positive Ulex europaeus agglutinin I staining. Upcyte mvECs also retained biological functionality such as tube formation, cell migration, and low density lipoprotein (LDL) uptake, which were still evident after PD27. Initial experiments using MTT and Live/Dead staining indicate that upcyte mvECs repopulate the BioVaSc Scaffold. As with conventional cultures, these cells also express key endothelial molecules (vWF, CD31, and eNOS) in a custom-made bioreactor system even after a prolonged period of 14 days. The combination of upcyte mvECs and the BioVaSc represents a novel and promising approach toward vascularizing bioreactor models which can better reflect organs, such as the liver.
Introduction
For our investigations we have used a decellularized porcine jejunal segment with an obtained vascular system called “BioVaSc” to establish a suitable attachment surface for EC culture. This model allows the development of an in vitro environment that, to a large extent, resembles intestinal physiology.18,33,34 Since the intestine is one of the most relevant organs for metabolic studies, reseeded intestine represents a cost- and time-efficient method that can be used to investigate toxic compounds. 34 The BioVaSc mainly consists of collagen I and is similar to the commercially available small intestine submucosa (SIS), a biocompatible framework that enables cellular migration and differentiation. 35 It has an arterial supply that allows the perfusion with fresh nutrients in a bioreactor system. Thus, the EC seeded on the vascular structures and lining the capillaries are provided with nutrients via the existing vascular network, and shear stress from the flow stimulates EC differentiation and function.
A big challenge in tissue engineering, in addition to ensuring the creation of the optimal microenvironment for cell growth and differentiation, is the isolation and expansion of appropriate cell types. 36 The yield of ECs is limited and often insufficient to re-populate medium to large biological scaffolds. The use of primary mvECs is hampered, as cells quickly dedifferentiate and obtaining donor tissue in sufficient amounts is difficult. 37 Furthermore, similar to all primary cells, mvECs have a limited lifespan, stop dividing, and become senescent after a finite number of cell divisions.38,39 To address this limitation, we explored the use of a novel technique that allows the generation of primary cell cultures with the ability to proliferate whilst maintaining their differentiated biological functionality. This so-called “upcyte” technology (derived from “upregulated primary cells”) involves the transduction of primary cells with proliferation-inducing genes without immortalization of the cells. 40 After transduction, the genetically modified cells are plated at low confluency to allow proliferation. Generally, the nontransduced cells die off or are diluted out over a number of passages. Once the transduced cells start proliferating, they can be passaged a number of times, depending on the cell type. Upcyte mvEC can be passaged for approximately 27 population doublings (PDs) compared with at most 12–15 PDs of primary mvECs, which means an additional 15 PD, that is, 3×104-fold more cells can be generated from one single donor. In addition, these cells are applicable for long-term studies. Therefore, upcyte mvECs may represent a suitable and extensive cell source for different applications, including the production of prevascularized scaffolds. Furthermore, upcyte mvECs have an important advantage over the use of human umbilical vein endothelial cells (HUVEC) for application in tissue engineering, because HUVECs are derived from fetal tissue and may, therefore, not represent a microvascular endothelium that is particularly involved in angiogenesis in vivo. 41 Transduced up-regulated primary mvECs can be used for engineering various kinds of prevascularized scaffolds for different applications such as bone grafts 37 and vascular grafts. 42 Moreover, the technology allows the generation of upcyte mvECs from various donors that are readily expanded to large quantities. We have already generated large batches of upcyte mvECs from more than six different donors. Here, we report the characterization of primary and upcyte mvECs. In order to investigate the effect of extended growth on the characteristics of the upcyte mvECs, the growth stages have been classified according to early (PD 5–10), mid (10–20), and late (PD 20–30) PDs. The characteristics tested included von Willebrandt Factor (vWF), CD31 and endothelial nitric oxide synthase (eNOS) expression, as well as biological functionality, which were demonstrated by their tube-formation capacity and low-density-lipoprotein (LDL) uptake. The EC marker, Ulex europaeus lectin I (UEA I), which recognizes different fucose residues in ECs, was also analyzed. Cell migration is another typical feature of mvECs. Primary mvECs stop dividing or de-differentiate at an early stage (P 2–3), 31 therefore, we determined whether the newly generated upcyte mvECs were able to undergo additional PDs while retaining their cell-specific properties. Here, we have used upcyte mvECs in proof-of-principle experiments to determine whether these cells can repopulate the decellularized matrix, the BioVaSc, in which complex capillary networks are formed.
Materials and Methods
Culture of primary and upcyte mvECs
Primary dermal mvECs (from human juvenile foreskin, Chair Tissue Engineering, and Regenerative Medicine, Universitätsklinikum Würzburg) and upcyte mvECs (Medicyte GmbH) were cultured in endothelial cell growth medium MV (ECGM; PromoCell) at 5% CO2 at 37°C. Cells were cultured as monolayers and had a doubling time of ∼65 h. The cells were passaged at 70–80% confluence. Briefly, the medium was aspirated, and the cells were washed with phosphate-buffered saline (PBS) for 10 min at 37°C. After removing PBS, trypsin/ethylenediaminetetraacetic acid (EDTA) (PAA) was added for 5 min at 37°C to detach the cells. The trypsin/EDTA reaction was stopped by the addition of ECGM (Promo Cell) (3:1 medium:trypsin). The trypsinized cells were transferred into a tube and centrifuged for 5 min at 60 g. The trypsinized cells were counted using Trypan Blue exclusion. For further culture, the cells were seeded at a density of 3000 cells/cm2.
Immunocytochemistry
Cells were washed with PBS and fixed with 100% methanol for 5 min at room temperature (RT). After fixation, the cells were rehydrated for 20 min using 3% bovine serum albumin (BSA)/PBS, which also inhibits nonspecific antibody binding. Each sample was incubated for 30 min at 37°C with the primary antibody diluted in blocking solution to detect CD31 (Dako; 1:300), vWF (Dako; 1:300), collagen I (Acris; 1:200), eNOS (Abcam; 1:100), CD105 (Dako; 1:50), CD34 (BioLegend; 1:100), CD146 (Abcam; 1:200), and Flk-1 (Santa Cruz; 1:200). After washing, the secondary antibodies Cy™3- conjugated AffiniPure goat anti-mouse immunoglobulin G (IgG) (1:200; Jackson Immuno Research) and Cy3-conjugated AffiniPure goat anti-rabbit IgG (1:200; Jackson Immuno Research) were incubated for 30 min at 37°C. Finally, the samples were washed three times with 0.2% BSA/PBS/0.05% Tween and stained with 4′,6-diamidino-2-phenylindol (DAPI) to detect nuclei. Marker presence was visualized by a fluorescence microscope (Axio Observer.Z1; Zeiss, objective Plan-Apochromat 20×, 0.8; software AxioVision 4.8.2).
Acetyl-LDL-uptake by ECs
Cultures of upcyte mvEC, primary mvEC, and HUVEC were plated onto eight-well Lab-Tek™ II Chamber Slides™ (Nunc) at a density of 10,000 cells/cm2 and cultured for 48 h at 5% CO2 and at 37°C. The medium was aspirated, and cells were incubated with 10–15 μg/mL 1,1-dioctadecyl-3,3,3′,3′-tertramethyl-indocarbocyanine perchlorate-labeled acetyl-LDL (Dil-Ac-LDL; Molecular Probes) for 4 h at 37°C to allow LDL uptake. The samples were washed once with PBS, fixed with ice-cold 70% ethanol/acetone (1:1) for 5 min, and then stained with DAPI/PBS (1 μg/mL) for 30 min at 37°C. The chamber slides were covered using a glass coverslip applying Mowiol (Sigma-Aldrich). LDL uptake was visualized using a fluorescence microscope.
Soft agar assay
A soft agar assay was used to measure the ability of cells to grow in an anchorage-independent manner. HepG2 cells were used as a positive control for anchorage independent growth. A volume of 50 μL 0.5% (w/v) agarose was added to each well of a 96-well plate. The agarose solution was prepared by mixing 1% (w/v) agarose solution and cell-culture medium (Dulbecco's Modified Eagle Medium (DMEM) supplemented with 20% fetal calf serum [FCS], 2 mM
Tube-formation assay
The tube-formation assay was used to monitor angiogenesis characteristics. The assay was adopted from Kubota et al. 43 Briefly, 100 μl Matrigel™ Basement Membrane Matrix (BD Biosciences) was added to wells in a precooled 96-well plate. The plate was incubated for 30 min at 37°C allowing the Matrigel to solidify. mvECs were diluted to different densities (4800, 6400, 9600, 12,800, 15,000 and 17,000 cells/cm2) and added to the matrix. The cells were incubated for approximately 24 h at 37°C until a tube network was formed. Pictures were taken after 2, 5, 8, and 24 h. HUVECs were used as a positive control.
Relative cell migration assay
The assay was performed according to Mastyugin et al. 28 . Primary and upcyte mvECs were cultured in supplemented ECGM (PromoCell) until they reached 50–70% confluence. HUVECs and primary mvECs were used as positive controls. Sixteen hours ahead of the experiment, cells were washed once with PBS and “starved” by culturing in serum-free ECGM containing 0.1% BSA (AppliChem). After the starvation period, cells were washed with PBS, carefully trypsinized with Accutase (PAA), and re-suspended in serum-free ECGM containing 0.1% BSA. The cell suspension, with different cell densities, was added to the top of each chamber in a final volume of 50 μL (96-well FluoroBlok™ Insert; BD Biosience), and the cells were allowed to attach for 1 h. The bottom chambers were filled with 200 μL of ECGM supplemented with 5% FBS, and the starved cells were allowed to migrate for 4 h. After cell migration, the cells were fixed by transferring the FluoroBlok (upper chamber) into a 96-well plate containing 200 μL 3.7% paraformaldehyde/PBS. After fixation, cells were washed with PBS and treated for 2 min with ice-cold methanol to permeabilize the cell membrane. Afterward, cells were rehydrated and washed with PBS before staining with 200 μL per well DAPI/PBS (1 μg/mL; Sigma) at RT for 10 min. Cells were then washed three times with PBS to remove unbound dye. Cells that migrated through the FluoroBlok membrane were visualized using a fluorescence microscope. All tested conditions were repeated at least in quadruplicate. Only the cells that migrated through the membrane are detectable, as the polyethylene terephthalate membrane effectively blocks the fluorescence signal from labeled cells that have not migrated. 28 The grade of cell migration in response to the chemoattractant, FCS, was measured by direct counting of the cells after staining the nuclei with DAPI.
Ulex europaeus agglutinin I staining
UEA I staining was carried out as decribed previously.44,45 Upcyte mvEC were seeded on to a 48-well plate and on reaching 70–80% confluence, they were fixed with 70% ethanol/acetone (1:1) for 5 min at RT. Thereafter, cells were washed three times with PBS (PAA) and incubated for 1 h with 1% BSA/PBS to block nonspecific binding sites. Then, cells were washed three times with PBS before incubation with Ulex europeaus agglutinin I lectin (Sigma) diluted by 1:50 in PBS (PAA) for 1 h at 37°C. Cells were co-stained with DAPI/PBS (1 μg/mL; Sigma) at RT for 30 min and washed thrice with PBS (PAA). Stained cells were visualized by fluorescence microscopy.
Porcine jejunal segment
A porcine jejunal segment for scaffold generation was obtained from a 3-month-old pig. All animals received human care in compliance with the Guide for care and Use of Laboratory Animal published by the National Institutes of Health (NIH publication No. 85-23, revised 1996) and the approval of our institutional animal protection board.
Decellularization of the porcine scaffold
The decellularization of the porcine scaffold was carried out according to our previously published method36,46 with minor modifications. After surgical removal, the segments were washed and stored overnight in PBS+ (Biochrom) with 1% gentamycin, streptomycin, and penicillin at 4°C. The next day, the arterial pedicle was connected to a bioreactor system and perfused with 500 mL 3% sodium desoxycholatmonohydrate solution for chemical decellularization (Roth). To protect the vascular system, the pressure was adjusted to 80 mmHg. Scaffold lumen was attached to a recirculating flow of 3% sodium desoxycholate until 500 mL of the solution had passed through the artery. The decellularized porcine jejunal segment was washed with 1000 mL PBS+ at a pressure of 100 mmHg to remove cell residues. The segments were stored overnight at 4°C in sodium desoxycholate. The next day, the segments were again connected to the bioreactor and were perfused with 2000 mL PBS+ to remove the sodium desoxycholate solution. The decellularized segment was then incubated with 150 mL DNAseI solution (60 mg in PBS+) (Roche) overnight at 4°C with 1% gentamycin, streptomycin, and penicillin. For removal of all chemical residues, the porcine scaffold was washed in PBS+ with antibiotics for 3 days. The solution was changed twice on the first day only. To sterilize the tissue, the scaffold was γ-irradiated with 25 kGy in PBS+ overnight (BBF Sterilizations service GmbH). To test the matrix for cell residues and DNA, segments of the cellular porcine jejunal matrix were incubated for 2 h in Histofix (4% paraformaldehyde) (Roth) and embedded in paraffin for histological analysis. Hematoxylin–eosin staining was performed according to standard protocols. The Feulgen reaction was used as a qualitative marker to detect DNA residues in the decellularized porcine tissue that was measured using a DNA staining kit (Merck).
Reseeding of the BioVaSc
The remaining vascular system within the decellularized scaffold was reseeded with 20–30 million cells upcyte mvECs in two sequential steps. In the first step, the scaffold was re-seeded through the arterial aditus and after a 2 h attachment period, cells were placed in the venous aditus. After the second seeding, the cells were allowed to attach for 2 h before the overnight pulsatile perfusion with 20 mmHg (amplitude±10 mmHg) was started. The next day, the perfusion pressure was increased over 7 h to 100 mmHg (amplitude±20 mmHg). The tissue culture was maintained in ECGM medium (Promocell) supplemented with 1% gentamycin and in a 5% CO2 atmosphere, and at 37°C for 14 days in a custom-made bioreactor system. 47 The medium was changed every 3–4 days. To ensure ideal culture conditions, temperature, gas exchange, pump activity (Ismatec), and systemic pressure levels in the bioreactor were computer controlled (Hewlett-Packhard; Software: Measure foundry, Data Translation) and adjusted when necessary.
Reseeding efficiency
To confirm the reseeding efficiency, Live-Dead staining and a vitality test were performed. For Live-Dead staining, a piece of the matrix was incubated in basal medium supplemented with 4 μL fluorescein diacetate and 25 μl propidium iodide for 7 min. The cells were then washed once with PBS+ and analyzed with a laser scanning microscope (Zeiss). For the viability test, a piece of the re-seeded scaffold was incubated for 3 h in medium with 1 mg/mL MTT (Invitrogen). The matrix was washed in PBS+ and analyzed under a standard light microscope.
Histological analysis
After 14 days, the reseeded BioVaSc was incubated for 2 h in Histofix and embedded in paraffin for histological and immunohistochemical analysis. Hematoxylin-eosin staining was performed according to standard protocols. For immunohistochemical characterization, the tissue was stained using the presence of CD31, eNOS, and vWF. Endogenous peroxidase and nonspecific binding was blocked with peroxidase blocking solution and antibody diluent, respectively (Dako). DuoVisonPlus with DAB Kit was used to detect the antibodies (DCS), and slides were counterstained with hematoxylin (Dako). Only samples with negative isotype controls were analyzed.
Results
Growth of mvECs
Figure 1 shows the growth of primary and upcyte mvECs over time. In contrast to primary mvECs that stopped growing after a few PDs (Fig. 1), upcyte mvECs were able to grow to a PD of 27 (Fig. 1). Thus, upcyte mvECs can achieve approximately 15 additional PDs, such that 3×104-fold more mvECs can be generated from a single donor. After PD 27, the cells stopped growing but did not die, evident as a plateau in the growth curve as well as the appearance of the senescence marker, senescence-associated β-galactosidase (data not shown). The upcyte mvECs could be expanded to generate significantly more cells compared with untreated cultures of primary mvECs.

Growth upcyte mvECs. The proliferation capacity of upcyte mvECs reached 27 PDs over 100 days. After PD 27, the upcyte mvECs stopped proliferating and reached senescence, evident as a plateau in the growth. mvECs, microvascular endothelial cells; PDs, population doublings. Color images available online at
Morphology
Upcyte and primary mvECs had similar morphologies and both proliferated at a rate of 2–5 PDs per week. Cells were grown as monolayers, during which time the cells were elongated. Upcyte mvECs grew well on plastic culture vessels and did not need a collagen matrix and, on reaching confluence, they exhibited the typical “cobblestone” appearance (Fig. 2).
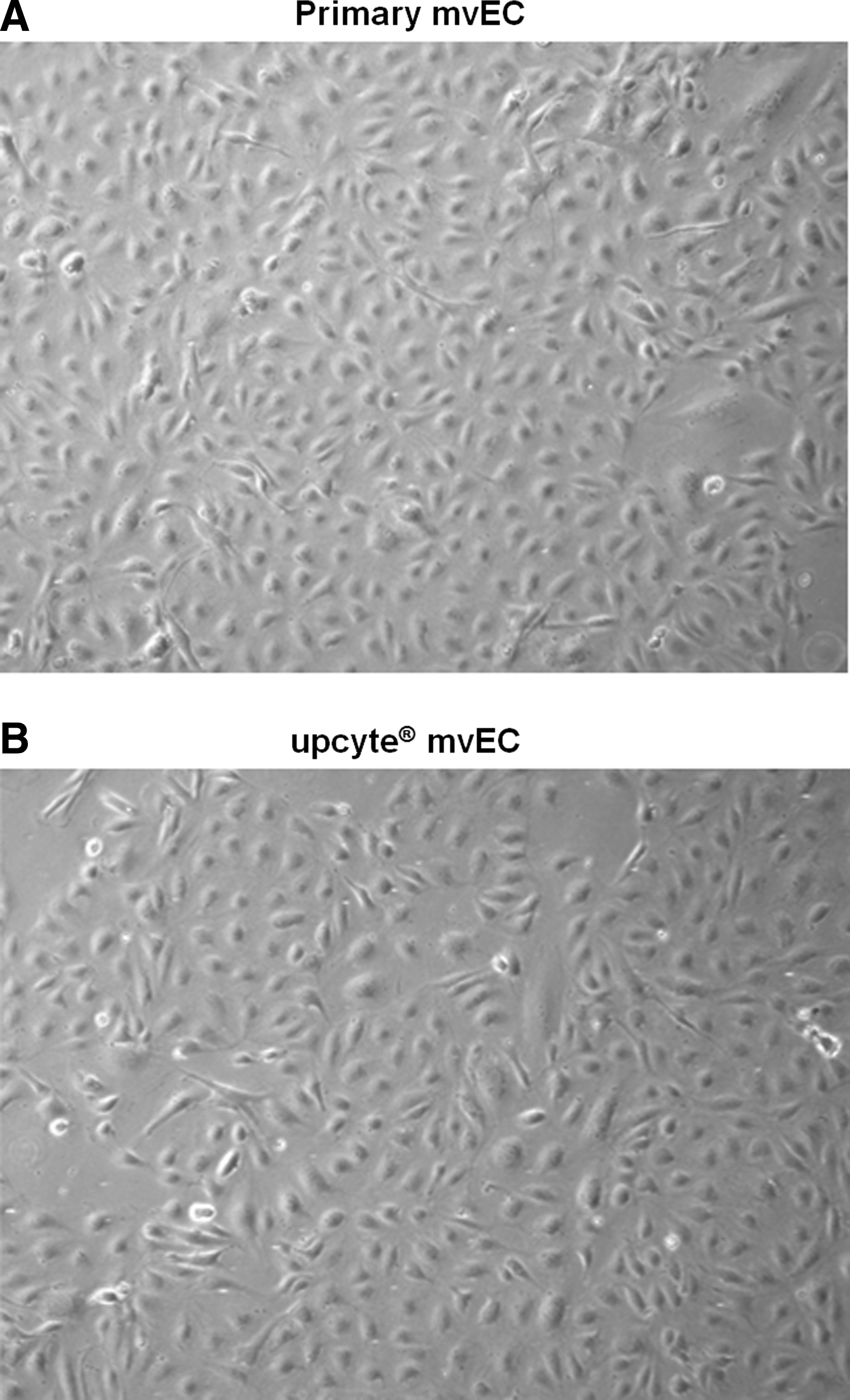
Light microscopy image of
Immunocytochemical characterization of mvECs
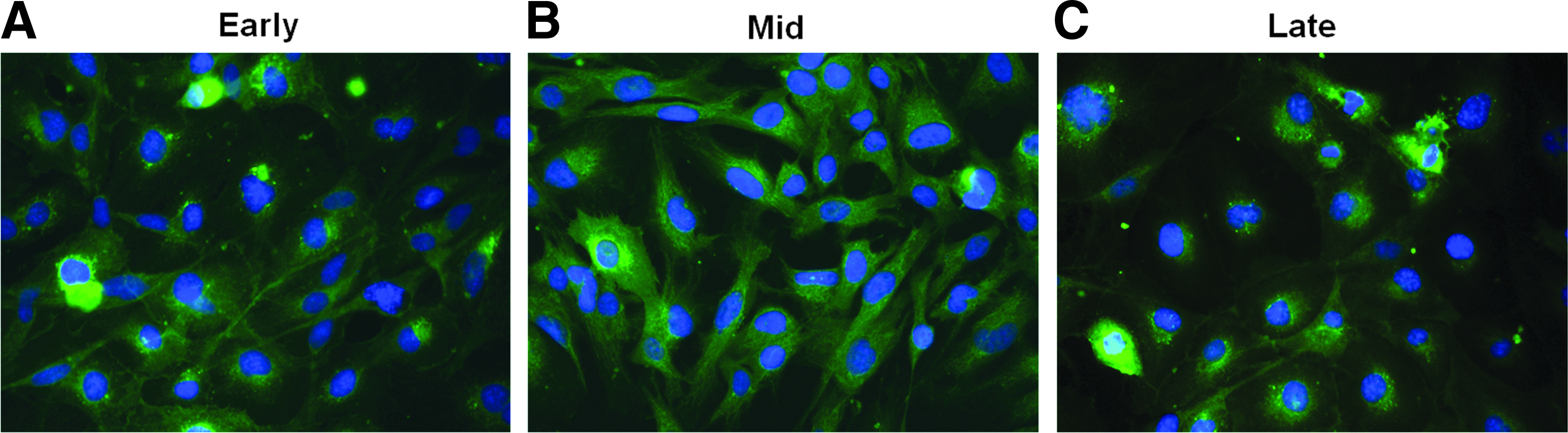
mvECs were characterized for cell-specific markers in early (PD 5–10), mid (PD 10–20), and late (PD 20–30) growth stages using immunofluorescence. All markers were expressed in comparable levels to primary mvECs. Upcyte mvECs clearly expressed the endothelial markers, CD31, vWF, and eNOS 48 in early, mid, and late PDs, respectively (Fig. 3). CD31 was expressed predominantly where cell contact was evident. UEA I was also analyzed in upcyte mvECs in early, mid, and late PDs. More than 95% of the cells were positive for fucose residues according to this marker (Fig. 4). ECs express little or no CD34 and collagen I; therefore, these markers were used as negative controls to exclude the presence of fibroblasts or other contaminating cells (data not shown). The expression of the differentiation marker, REIC/Dkk3, in upcyte mvECs was analyzed by real time quantitative PCR. 48 The level of REIC/DKK3 mRNA, in mvECs from all PDs tested was equivalent to that of primary mvECs (Fig. 5).

Immunohistological analysis of the expression of different EC-specific markers in upcyte mvECs in early [PD 7–12
UEA I lectin in early [PD 5.4

Expression of the differentiation marker, REIC/Dkk3, in upcyte mvECs in early [PD 5.4], mid [PD 14.1], and late expression [PD 24.4]. The data are expressed as a ratio of the levels in primary mvECs (all normalized to GAPDH). The level of REIC/DKK3 mRNA, in mvECs from all PDs tested, was equivalent to that of primary mvECs.
Soft agar colony formation assay
Upcyte mvECs (PD 23.4) were anchorage dependent, as they did not form colonies in soft agar and remained as single cells after 7 days in culture. By contrast, over the same time, HepG2 cells (P3), which grow anchorage independently,49,50 formed colonies in soft agar.
Dil-Ac-LDL uptake
Uptake of Dil-Ac-LDL was clearly demonstrated for both primary (PD 7) and upcyte mvECs (PD 21) (Fig. 6). Almost 98% of the cells showed a positive staining, and only low background fluorescence was observed. The bright staining of Dil-Ac-LDL in the cells showed a typical spotty perinuclear distribution.

Dil-Ac-LDL labeling of
Cell tube formation
Upcyte mvECs (PD 19) formed capillary-like structures which were comparable to that demonstrated by HUVEC (PD 17.2) and primary mvECs (PD 9) (Fig. 7). The cells started forming capillary-like structures within 2 h and had formed a network of capillaries by 8 h. The network presented a central lumen surrounded by ECs. Different donors were tested, and all donors built a typical network of capillary-like structures (data not shown).

Tubule formation by
Relative cell migration
The migration of mvECs from different donors through basement membranes was monitored. Upcyte mvECs (PD 18) displayed the ability to migrate, and their responses to FCS were similar to those of the two positive controls, HUVECs (PD 16) and primary mvECs (PD 5) (Fig. 8).The grade of migration of upcyte mvECs was higher than that for primary mvECs (which were from different donors).

Cell migration of
Generation of a decellularized biological scaffold and characterization of the reseeded BioVaSc
Histological analysis confirmed the decellularization of the porcine jejunal segment, including its feeding artery and draining vein. A few cellular and DNA residues, detected using hematoxylin-eosin staining and a Feulgen reaction, were evident by the end of the treatment.
The injected cells that repopulated the vascular structure were all prescreened to confirm the expression of the characteristic vascular surface markers, CD31, vWF, and eNOS (Fig. 9A). The reseeding of the blood vessels with upcyte mvECs (PD 15–19) was visualized using the Live/Dead and MTT assays. For Live/Dead staining, fluorescein diacetate and Propidium iodide was used. Fluorescein diacetate is transformed through living cells into fluorescein, which shows green fluorescence. Propidium iodide goes through defect cell membranes and interacts with DNA, so dead cells show red fluorescence. The Live/Dead assay was evaluated with a Laser Scanning Microscope (Zeiss; LSM 710), and there was clear evidence that the blood vessels were reseeded with living cells (green) and only a few dead cells (red) were visible. Furthermore, the cells line the branches of the vessels (Fig. 9C). The viability of the mvECs was also confirmed using MTT. Living cells reduce the yellow MTT into blue formazan and appear blue. This experiment was evaluated by light microscopy through the matrix (Nikon; Eclipse TS 100-F) and also shows reseeding of the blood vessels and branches with upcyte mvEC (Fig. 9D). Therefore, the outcomes from the Live/Dead staining are approved.
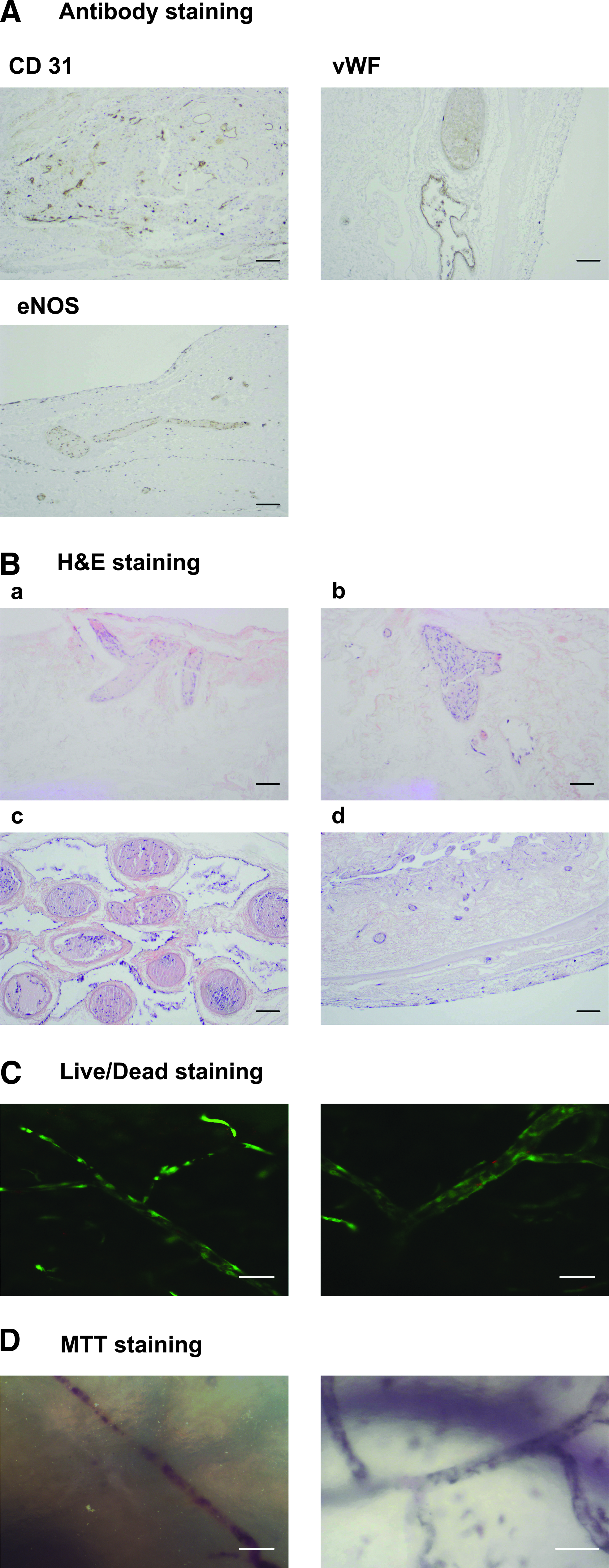
A histological analysis of cross-sections through the matrix with hematoxylin-eosin staining shows cells in blue and the matrix in red. Analyses with light microscopy (Zeiss; AxioVert 200M) show that cells are present in the blood vessels (Fig. 9B[a–c]) and can also be found in the lumen of the gut and migrated inside the matrix (Fig. 9B[d]).
Discussion
Tissue engineering was developed as an alternative to generate transplants for the treatment of tissue loss or end-stage organ failure, resolving the shortage in organs.51,52 The new technologies established in material research and the cell-culture technique has led to the development of long-time bioartificial tissues that can be used as models for experimental setups reflecting underlying physiological mechanisms in the human body.53,54 The supply of nutrients to the construct is important to keep the grafts viable. Tissues sections that are approximately 200 μm thick can be supplied by diffusion alone.55,56 For thicker tissues, diffusion is not sufficient,57–59 and vascularization or a similar mechanism such as a hollow fiber-based perfusion system is necessary. 60 It is also essential to ensure the functionality of cultured cells; therefore, models are needed that mimic the microenvironment, such as advanced bioreactor technologies and co-culture of different cell types. 61
The BioVaSc is a collagen-based network, similar to the commercially available SIS. It is a biocompatible framework and enables cellular migration and differentiation. 35 We have optimized the perfusion parameters using systematic mathematical modeling and computer simulation such that the natural environment of the body could be mimicked, from blood pressure to temperature. 36 The combination of a prevascularized scaffold and a tissue-specific bioreactor should provide a sufficient nutrient supply to complex 3D bioartificial tissues. The reseeded capillary network of the BioVaSc can be used for the production of various test tissues, for example, multiple hepatocyten layers. 19
A limitation of primary mvECs is that the typical yield from a single isolation is not sufficient to re-populate a prevascularized scaffold. Primary mvECs have a limited lifespan and stop dividing after a finite number of cell divisions. Moreover, primary mvECs start dedifferentiating within a few days. This problem has been addressed by developing upcyte mvECs that are able to undergo multiple cellular divisions—up to 27 PDs—while retaining the phenotype of primary mvECs. In addition, large batches of upcyte mvECs can be generated from different donors and can be used for different applications in tissue engineering such as bone grafts 37 or vascular grafts. 42
Here, we have demonstrated that upcyte mvECs can be compared very well with primary mvECs in terms of morphology, cell-specific markers, and biological functionality. As for primary cells, newly generated upcyte mvECs retain the typical “cobblestone” morphology when they are at confluency.
62
The glycoproteins, vWF, and CD31, are produced predominantly by ECs,
63
and CD31 is enriched in the intracellular junctional domains of cultured ECs.64,65 Likewise, these two glycoproteins were clearly detected in upcyte mvECs, and were concentrated in areas of cell–cell contact in cell monolayers. Another identifying feature of ECs is UEA I binding to alpha-
In addition to cell-specific markers, upcyte mvECs should also be expected to have functional characteristics of primary cells, such as LDL uptake, tube formation, and cell migration. Primary and upcyte mvEC were able to take up LDL, and nearly 100% of the cells in the culture were shown to exhibit this function. The ability of cells to form tubes is a defining feature of EC populations, 27 which rapidly form capillary-like structures in vitro when they are plated on top of a basement membrane extracellular matrix. 66 This process includes all the important steps in angiogenesis, including mvEC adhesion, migration, alignment, and tube formation.27,28 We found that upcyte mvECs were able to form tubes and, moreover, the grade of their migration was higher than that for primary mvECs. However, this difference in migration efficiency could be due to the fact that the upcyte and primary mvECs isolated were from different donors.
Having confirmed the differentiated status of the upcyte mvECs, we then explored whether they could be used in the BioVaSc. Data presented here confirm that the method we employed to decellularize the tissue and produce the matrix was free of both cell and DNA residues. The reseeding of the matrix was monitored using immunofluorescence staining of the cells. The mvECs lining the vessel structures were endothelial in nature and showed the typical markers, CD 31, vWF, and eNOS. We also confirmed that the majority of cells which were reseeded were viable, using MTT metabolism and Live/Dead staining.
The feasibility of repopulating vessel bed structures was first shown in 2005, 46 but an incomplete lining of the vessels was observed. To enhance the reseeding, vessel lumens were exposed to CCN1, which has been shown to support the repopulating process. 21 The experiments showed that a higher reseeding efficiency was possible with human cord blood-derived ECs. However, complete reendothelialization did not occur. Therefore, our experiments aimed at reseeding the BioVaSc with sufficient numbers of mvECs.
We were able to show that the upcyte mvECs reseeded the matrix, but the reendothelialization was not complete. Future experiments could investigate whether CCN1 could improve the process further.
In summary, we report a novel and promising system for the generation of a perfusable, endothelialized matrix for artificial tissues in vitro. Moreover, upcyte mvECs were shown to be comparable to primary mvECs and, thus, represent a sufficient and reliable alternative to primary mvECs to generate prevascularized scaffolds. This matrix comprised a reseeded vascular structure in which the lumen of the scaffold could, in addition to mvECs, be reseeded with cells from liver or intestine (e.g., Caco-2).3,18 The resulting complex and multi-cellular human tissue models would be useful alternatives to animal experiments for measuring drug penetration, distribution, and metabolism. 3
Footnotes
Acknowledgments
The research leading to these results has received funding from the European Union's Seventh Framework Program (FP7/2007–2013) under Grant Agreement no 242175. The authors thank Nicola Hewitt for critical review and editing of the article. The authors want to thank Dr. Martin Schenk from the experimental medicine of the University of Tübingen for making the operating room available and Dominique Tordy for the preparation of the matrices.
Disclosure Statement
The following authors have an employment position to disclose: Katharina Scheller, Nadja Hartmann, Bernhard Münst, and Joris Braspenning hold a full-time employment for an entity having a commercial interest in the subject matter under consideration in the article. Name of entity: Medicyte GmbH. Upcyte® is a registrated trademark of Medicyte GmbH.