
Abstract
Irradiated head and neck cancer patients suffer from irreversible loss of salivary gland (SG) function, along with significant morbidity and compromised quality of life. To date there is no biologically-based treatment for this distress. Adult salivary gland stem cells are promising candidates for autologous transplantation therapy in the context of tissue-engineered artificial SGs or direct cell therapy. The major restrictions in handling such cells are their limited lifespan during in vitro cultivation, resulting in a narrow time-window for implantation and a risk of tumorigenic changes during culture. To overcome these difficulties, we tested in a rat model the possibility of establishing a personal/autologous SG stem cell bank. SG's integrin-α6β1-expressing cells were shown to hold a subpopulation of SG-specific progenitor-cells. Explanted and cultured single cell-originated clones were cryopreserved for up to 3 years and shown to exhibit genetic and functional stability similar to noncryopreserved cells, as was emphasized by soft agar assay, division potential assessment, flow cytometric analysis, real-time reverse transcriptase–polymerase chain reaction, in vitro three-dimensional differentiation assay, and immunofluorescence confocal microscopy. Future integration of the novel strategies presented herein to a clinical therapeutic model will allow safe preservation until transplantation and repeated transplantation if needed. These tools open a new venue for adult autologous stem-cell transplantation-based SG regeneration.
Introduction
To overcome these difficulties, the aim of this study was to test the concept of establishing a personal/autologous SG single cell-originated stem cell bank. 9 This cell banking system could allow multiple and prolonged autologous therapies upon necessity. SGIE were cryopreserved for up to 3 years and evaluated for genetic and functional stability by soft agar assay, fluorescence-activated cell sorting (FACS), differentiating culture, confocal immunofluorescence, and TaqMan real-time polymerase chain reaction (PCR). Further, as these cells were preserved for long periods of time, there was increased risk for tumorigenic changes and stronger possibility of other cells from different lineages unrelated to the target population to reproduce during re-cultivation. To reduce these obstacles, we developed a methodology for the production of single cell-based salivary stem-cell clones. This methodology opens a window for future clinical implantation safety assurance.
Materials and Methods
Animals
Male Sprague-Dawley rats (200–300 g) were used as the animal model. All animal treatment procedures were approved by the Animal Care and Use Committee at our institute and animals were continuously monitored for any signs of distress. They were kept at 22°C±2°C, and were euthanized with IP injection of veterinary Pental (pentobarbitone sodium, 200 mg/mL; doze: 1 mL/1.5 kg body weight, according to the manufacturer instructions; CTS Chemical Industries Ltd.).
Immunomagnetic isolation of integrin α6β1-positive cells
Isolation of SG stem cells was performed as we previously described.2,4,10
Cultivation of integrin α6β1-positive (SGIE) cells and murine prostate cancer PC3 cells
SGIE culture
The isolated cells were inoculated into 25-cm2 Falcon flasks preseeded with 2×105 lethally irradiated (50 Gy) NIH 3T3 mouse fibroblasts serving as a feeder layer, as previously described, or into flasks precoated with 10 μg/cm2 of rat collagen type I (Sigma). Parenchymal cells were maintained in a 3:1 mixture of Dulbecco's modified Eagle's medium (DMEM) and Ham's F-12 supplemented with 2 mM glutamine, 100 units/mL penicillin, 100 units/mL streptomycin, and 0.25 μg/mL amphotericin (Biological Industries). The medium was supplemented with epidermal growth factor (10 ng/mL), insulin (5 μg/mL), transferrin (5 μg/mL), triiodothyronine (T3; 2×10−9M), hydrocortisone (0.4 μg/mL), adenine (1.8×10−4M), and cholera toxin (10×10−10 M) (Sigma). When cells reached 80% confluence, they were released with 0.25% (w/v) trypsin–0.05% (w/v) ethylenediaminetetraacetic acid (EDTA) solution (Biological Industries), centrifuged at room temperature (1300 g), and counted with a Cellometer™ Auto T4 cell counter (Nexcelom). About 5×105 cells were plated for continuous culture as previously described. The remaining cells were used for further analysis.
PC3 culture
PC3, a murine prostate cancer cell line, was a generous gift from the Laboratory of Experimental Surgery, Hadassah University Hospital, Jerusalem. Cells were grown on DMEM supplemented with 10% FCS, 2 mM glutamine, 100 units/mL penicillin, 100 units/mL streptomycin, and 0.25 mg/mL amphotericin (Biological Industries). At 80% confluence, PC3 cells were passaged with trypsin-EDTA solution as described for SGIE.
Limiting dilution assay and cryopreservation
To isolate colonies derived from a single cell, a limiting dilution assay was utilized as follows: a unique conditioned medium was prepared from two parts fresh growth medium, one part 2-day-used SGIE growth medium and one part used irradiated NIH 3T3 medium (2:1:1, v/v). A limited number of cells from passage 6–8 were diluted to a concentration of 0.5 cells per 200 μL of conditioned growth medium, and then plated in a 96-well plate precoated with collagen type I from rat tail (Sigma). The plates were monitored daily under an inverted microscope (model IX71; Olympus Corporation), and photographs were taken with a CFW-1310C camera (Scion Corporation). For negative control, SGIE were diluted to the same concentration and were cultured on 96-well plates with unconditioned medium as mentioned above. After 7 days, the obtained clones were released using trypsin-EDTA solution and transferred to a collagen I precoated 24-well culture dish. In the following passages, the clones were transferred to a 6-well culture dish and then to a 25-cm2 culture flask. Only clones that were culture on 25-cm2 flasks were considered positive for the clones counting procedure.
The cryopreservation procedure was performed as previously described 11 with modifications. For cryopreservation, 1×106 SGIE cells originated from single cells were suspended in 1.5 mL growth medium supplemented with 10% (v/v) dimethyl sulfoxide (Sigma) in Nunc CryoTubes. The CryoTubes were rapidly transferred to a Tarsons Cryocooler and placed in −80C° for 24 h. When loaded with 250 mL isopropyl alcohol (Gadot Ltd.) the Tarsons Cryocooler provides a gradual cooling rate of −1C°/1 min as it is placed in a −80C° freezer. 12 The CryoTubes were then transferred to a liquid nitrogen container for up to 3 years.
Thawing, viability assay, and reculture of cryopreserved cells
Cryopreserved cells were rapidly thawed in a 37°C water bath 9 and then centrifuged at room temperature (1300 g). The pellet was resuspended in fresh growth medium as described above. To test cells viability, Trypan blue dye exclusion assay was performed as previously described 13 with modifications. Thawed SGIE clones were centrifuged again (3 min, 1300 g) and suspended in 1 mL PBS ×1 (Biological Industries). Thirty microliters of the cell suspension were mixed with an equal volume of 0.4% Trypan blue stain (Molecular Probes) and were allowed to stand for 5 min at room temperature. Twenty microliters of the suspended cells were counted with Cellometer™ Auto T4 cell counter as described above, excluding the dead stained cells.
For further studies, about 1.5×106 cells were seeded in collagen I-coated 25-cm2 flasks.
Cells counting for division estimation
About 5×104 to 10×104 cells of either cryopreserved or noncryopreseved (nCP) cell cultures were seeded on 25-cm2 flasks and were counted as we and others previously described.2,10,14
Soft agar assay
Soft agar assay was performed as we and others previously described.10,15
FACS analysis
Flow cytometric analysis was performed in a C6 Flow Cytometer according to the manufacturer's instructions (Accuri Cytometers). Briefly, cells were divided into two aliquots and washed with PBS. One aliquot of the cells was further incubated with rat integrin α6β1 IgG1 monoclonal antibody (1:100 v/v, Chemicon) and with rabbit anti-c-KIT (1:100 v/v in 1.25% FCS in PBS, CD117; Santa Cruz Biotechnology) for 30 min at room temperature. Then the cells were incubated for 30 min with secondary antibody, Alexa Fluor 488-conjugated goat anti-mouse IgG or Alexa 647-conjugated donkey anti-rabbit antibody (both 1:100 v/v in 1.25% FCS in PBS; Invitrogen). As a control, the second aliquot of cells was incubated with the secondary antibodies only.
Three-dimensional cultures
Three-dimensional (3D) Matrigel™ cultures were prepared as described previously. 10 Briefly, growth factor reduced Matrigel or basement membrane Matrigel (200 μL; Becton Dickinson) was added to 24-well tissue culture dishes and incubated for at least 30 min at 37°C to polymerize. SGIE were plated at a density of 1.5×105 cells/well and were cultured in a 37°C, 5% CO2 incubator for 3 days. Cells were photographed under an Olympus inverted fluorescent microscope (model IX71) with a CFW-1310C camera.
Immunofluorescence and confocal microscopy
Immunofluorescence assay was performed as we previously described 10 with the following modifications: The antibodies against amylase (2 μg/mL, mouse monoclonal IgG; Santa Cruz Biotechnology), p63 (200 mg/mL, rabbit polyclonal IgG; Santa Cruz Biotechnology), cystatin C (1 μg/mL, rabbit polyclonal IgG; Chemicon), integrin-α6β1 (mouse monoclonal IgG; Chemicon), c-KIT (CD117, 2 μg/mL; Santa Cruz), and CK17 (Rabbit Polyclonal; Epitomics) were diluted 1:100 with 1.25% FCS in PBS and incubated overnight at 4°C. Alexa 488- and Alexa 647-conjugated secondary antibodies against rabbit and mouse IgG respectively (Molecular Probes) were added to samples at a dilution of 1:500 and incubated for 90 min at room temperature. For nuclear staining, 40,6-diamidino-2-phenylindole (DAPI, 1 μg/mL; KPL) was added to samples and incubated for 10 min at room temperature. The stained preparations were evaluated using a confocal laser scanning microscope (Model 710; Zeiss).
RNA isolation and real-time PCR analysis
RNA was extracted from 1×106 SGIE that had been recultured for one to two passages after 2 years of cryopreservation (n=5, passages 10–13). nCP SGIE (1×106) served as the control group (n=3, passages 6–7). All samples were cultured in collagen I-coated 25-cm2 flasks. Extraction of total RNA and synthesis of complementary DNA were performed using TaqMan Cells to CT kit (Applied Biosystems) according to the manufacturer's instructions, as previously described. 10 To extract RNA from 3D cultures, 24-well Matrigel-cultured cells were incubated with 500 μL dispase solution (Becton Dickinson) for 2 h in a 37°C, 5% CO2 incubator. The depolarized mixture of cells, Matrigel and dispase was collected into 15-mL tubes (contents of six to nine wells per tube) and centrifuged at 4°C (5 min, 1300 g). The pellet was washed twice with cold PBS. Total RNA extraction and synthesis of cDNA were performed as described above. cDNA was amplified using the 7300 real-time PCR system (Applied Biosystems) with TaqMan Gene Expression Assay for α-amylase, cystatin C, CK17, AQP5, and glyceraldehyde 3-phophate dehydrogenase (GAPDH; Applied Biosystems).
Statistical analysis
The results were statistically analyzed by paired Student's t-test, with p<0.05 considered statistically significant. All the data presented here indicate mean±SEM, unless otherwise stated.
Results
Preparation and cryopreservation and tumorigenic potential of single cell-originated clones
SGIE were immunomagnetically isolated from rat submandibular SGs. In each cell preparation, about 1×106 cells were typically isolated and grown in separate collagen-coated culture dishes until passage 6–8. To isolate colonies derived from a single cell, limiting dilution assays were performed. Isolated (single) cells that were cultured with the unconditioned medium adhered to the surface of the plate, but could not reproduce to form clones (Fig. 1a, arrows). Cells that were cultured with the conditioned medium started to divide within 2 days after plating, and formed mature clones within 7–10 days (Fig. 1b, arrows). The percentage clone yield in the conditioned media culture was 4.239%±0.988% (Fig. 1c, n=29.). Conversely, cells cultured with unconditioned medium, produced no clones (Fig. 1c, n=10). These clones were cultured for additional two to four passages and cryopreserved for up to 3 years. After the specified periods of time, cells were thawed and their function was examined. Immediately after thawing, Trypan blue dye exclusion assay revealed that 70.2%±5.26% were viable after 3 years of cryopreservation (n=5). To test the cells' ability to form colonies under anchorage-independent conditions in a semi-solid medium, a soft agar assay was carried out. SGIE (before and after cryopreservation) and 1×105 PC3 cells, serving as a positive control, were separately integrated into a liquid agar medium mixture and seeded on top of a base layer for 6 days. SGIE did not produce colonies on the soft agar either before (Fig. 1d, I) or after (Fig. 1d, II) cryopreservation, whereas the PC3 cells did (Fig. 1d, III arrows).

Isolation of single cell-originated clones by limiting dilution. No cell division was observed during the 5 days duration of cultivation using unconditioned medium
Stem cell marker expression and division numbers stability of cryopreserved SGIE
To examine whether the SGIE maintain stem cell marker stability after long cryopreservation, thawing, and recultivation, we examined the expression of integrin α6β1 and c-KIT by flow cytometric analysis. In cultures based on integrin α6β1 isolation, 92.3±1.67% of nCP cells (passages 2–4, n=4) was positive to integrin α6β1. After 2 years of cryopreservation 95%±0.69% of the cells (CP, passages 10–14, n=4) remained positive to α6β1 (Fig. 2a, R4). In both nCP and CP populations, three different c-KIT-expressing cell subpopulations were observed. The majority of the cell population demonstrated low percentage of c-KIT expression (Fig. 2a, R1; 83.58%±3.53% and 84.83%±2.53%, respectively), followed by an intermediate c-KIT and a high c-KIT-expressing cell population (Fig. 2a, R2; nCP: 3.026%±0.56%, CP: 3.97%±0.59%, R3; nCP: 5.7%±1.4%, CP 6.21%±2.19%, respectively). No statistically significant differences were found between nCP and CP samples either in integrin α6β1 or in c-KIT expression levels (p>0.26 for all gates).

Expression of stem cell markers and division capability before and after cryopreservation. Representative flow cytometric analysis shows similar high α6β1 expression in both nCP and CP cultures (R4, 92.3±1.67 and 95±0.69%, respectively. p=0.44). R1: low-c-KIT-expressing cells (83.58%±3.53% and 84.83%±2.53%, respectively, p=0.81). R2: intermediate-c-KIT-expressing cells (3.026±0.56 and 3.97±0.59, respectively, p=0.26).R3: high-c-KIT-expressing cells: 5.7%±1.4% and 6.21%±2.19%, respectively, p=0.85). n=4 for both cultures in all fluorescence-activated cell sorting experiments
Cultures of nCP and 1–3 years CP SGIE were further estimated for division number assays (Fig. 2b, c; passages 10–13). No differences were found when comparing the averages of both the division numbers within each passage (Fig. 2b) and total division numbers (Fig. 2c; p>0.3 for all passages and for overall estimation).
Structure formation, protein and gene expression stability of cryopreserved SGIE
nCP and CP after 3-years (time limit of the experiment) SGIE were cultured on two-dimensional collagen I (Fig. 3a) and 3D Matrigel scaffolds (Fig. 3b) for 3 days with serum-free medium. Cultures on collagen I matrix, formed SGIE clones lacking any definite structures. Conversely, upon culturing on 100% Matrigel, net-like structures composed of duct-like branches terminating with spheres were established.

Morphological organization, protein expression, and mRNA expression. nCP (passage 14) and CP (passage 9) SGIE clones cultured on collagen I
Immunofluorescence and confocal microscopy of SGIE after 3 years of cryopreservation grown on collagen type I for 3 days revealed expression of the membrane integrin α6β1 protein and the nuclear transcription factor p63 (Fig. 3c),and the presence of the cytoplasmic salivary-specific protein markers α-amylase and cystatin C (Fig. 3d).Conversely, upon cultivation on Matrigel for 3 days, SGIE created acinar-like structures and expression of both α6β1 and p63 proteins was largely reduced (Fig. 3e), along with decreased expression of α-amylase, with the exception of the structural margins where cells maintained high expression of α-amylase and cystatin C (Fig. 3f)
A comparison of mRNA expression profiles of nCP (passage 6–7) and 3-year-CP recultured (passages 10–14) SGIE cultured in a collagen I-coated plastic flask revealed no significant changes in the mRNA expression levels of aquaporin 5 (AQP5), CK17 or cystatin C (Fig. 3g). In contrast, a 6.8-fold decrease in α-amylase mRNA expression was observed in the cryopreserved cells.
Discussion
In the process of developing any clinical application of cell therapy, it is essential to perform detailed and careful studies of cell longevity and cell safety. In this context, the purpose of the present study was to evaluate these parameters in animals as a model for future use in human clinical trials toward re-engineering radiation-induced SG hypofunction. The present study was conducted with the aid of the relevant “Guidelines for the Clinical Translation of Stem Cells” established by the “International Society for Stem Cell Research 2008 (ISSCR 2008),” specifically, those concerned with (1) storage and cell banking and (2) the risk of tumorigenicity during culture. This is the first study to implement the strategy of SG-originated stem cell cryopreservation for future clinical application to regenerate SG in head- and neck-irradiated patients in the context of tissue engineered artificial SGs or direct cell therapy.
Stem cell storage and banking are fundamental issues in tissue repair, as it has been suggested for several epithelial-originated organs such as human limbal epithelial stem cells, 16 conjunctival epithelial stem cells, 17 pancreatic ductal cells, 18 and hepatocytes. 19 Human hepatocytes do not tolerate cryopreservation, as reflected by impairment of cell adhesion, morphological changes in mitochondria, loss of ATP production, alteration in mitochondrial respiratory chain enzymes, increased mitochondrial permeability, and loss of membrane potential. 20 Nevertheless, viability test of cryopreserved SG in this study exhibited a good percentage of recovery cells in agreement with recovery of cryopreserved human limbal epithelial stem cells and conjunctival epithelial stem cells (∼70%–80%)16,17 In SGs, personal stem cell banking may be important in the repair process by autologous transplantation after IR for three reasons: transplantationcan only be performed after irradiation treatment, which lasts 2 months. This time frame might result in reduced transplantation efficiency due to a decrease in cell-specific fraction numbers, as has been reported for c-KIT-expressing salivary stem cells. 3 Second, under normal circumstances transplantation success can be limited (for instance, 69% using c-KIT-expressing cells), 3 and thus more than one transplantation procedure may be required for successful treatment. Third, possible alterations in cell function long after transplantation might require additional transplantation therapy in some patients (Fig. 4). A first step to achieving safe long-term preservation of cells is the ability to produce single cell-based clones. We succeeded in producing such clones by limiting dilution assay employing a unique conditioned medium. In previous studies, SG clones were produced as multiple separate clones in a dish.2,21 The advantage of the methodology suggested herein lies not only in obtaining scientifically reproducible results, but also, and more importantly, in its providing a safety measure toward future clinical application: using cultures derived from a single cell, rather than cells originated from a mixture of cell clones, enables better evaluation of whether a representative cell sample is expressing a tumor-related phenotype. To further analyze the safety issue, we analyzed whether the long-term cryopreservation process causes changes in a soft agar test. Consequently, no alterations were found, indicating no tumorigenic changes, due to the cryopreservation-thawing-reculturing process. These findings are in agreement with previous studies indicating that cryopreservation does not alter the tumorigenic characteristics of stem/progenitor cells derived from mesenchymal tissues such as the periodontal ligament, 22 peripheral blood, 23 and dental apical papilla. 24 We next evaluated whether the expression of stem cell membrane markers are altered as a result of the cryopreservation process and indeed, integrin α6β1 and c-KIT expression showed similar expression pattern. Integrin α6β1 protein markers are used for cell isolation 2 as they indicate the cell's origin from the basal ductal area. 25 c-KIT (CD117) is a type III receptor tyrosine kinase that operates in cell-signal transduction in several cell types. Normally, c-KIT is activated (phosphorylated) by binding to its ligand, stem cell factor. 26 Previous studies have demonstrated that hepatic oval cells express both c-KIT and stem cell factor. 27 More recently, it has been shown that FACS-isolated Sca-1/c-KIT-expressing mouse SG cells transdifferentiate into pancreas and liver lineages. 28 Further, injection of irradiated mice with only 300 to 1,000 c-KIT-expressing cells isolated from cultured salispheres resulted in markedly improved saliva secretion. 3 However, the c-KIT stem cell pool in the salispheres remains heterogeneous and culture spheres from the single-cell suspensions are not re-culturable. Moreover, after 10 days of culture, c-KIT expression in the salispheres is confined to their periphery. Hence, only cells that are cultured for fewer days are suitable for implantation.
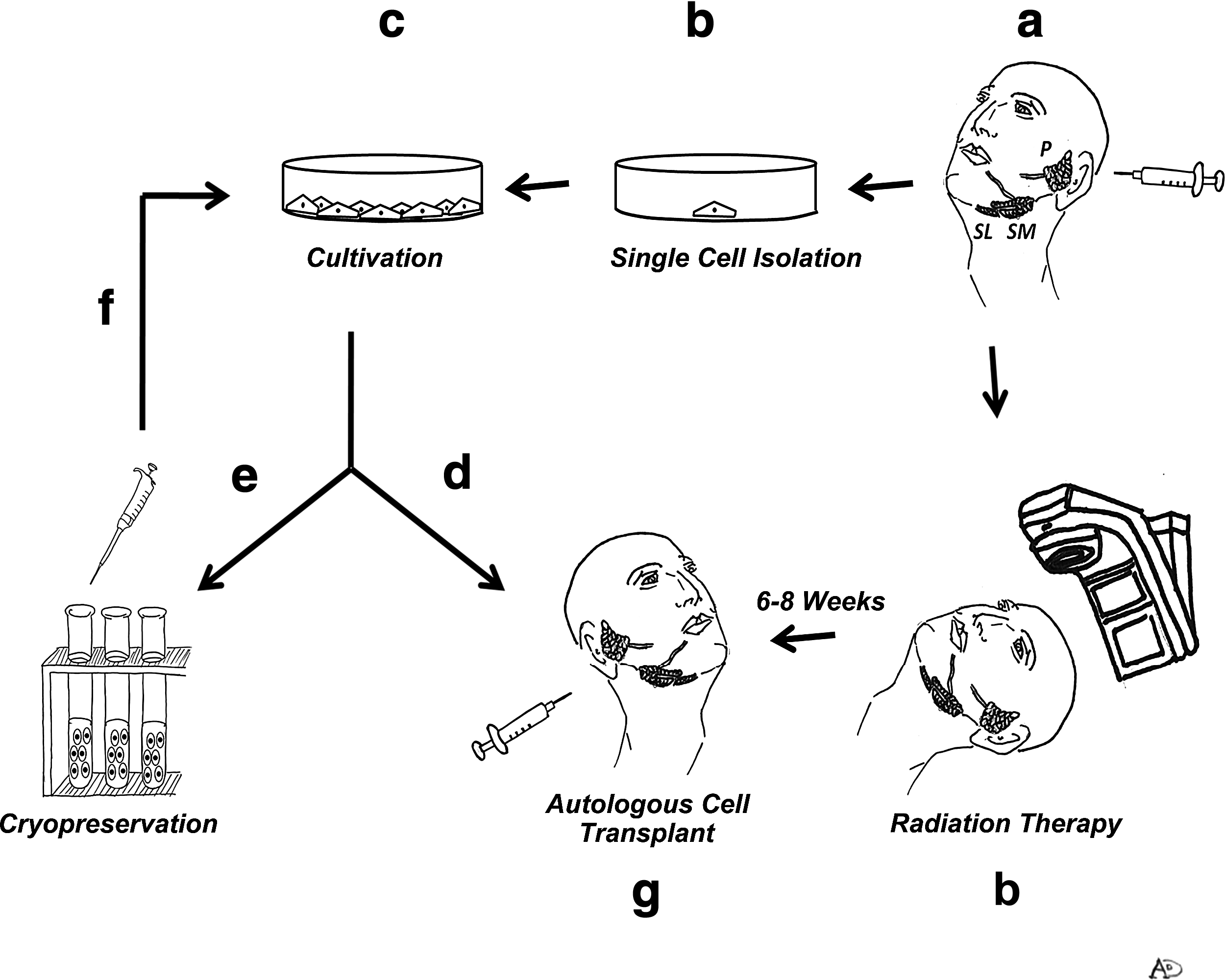
Suggested human model for autologous transplantation of CP SGIE. Upon diagnosis of head or neck cancer, SGIE from the unaffected major salivary glands are isolated
Three distinct c-KIT-expressing subpopulations of cells were observed in the enriched α6β1-expressing cells population in both non- and cryopreserved conditions.6% of SGIE population corresponded to high c-KIT-expressing cells, followed by ∼3 to 4% intermediate c-KIT-expressing cells, and ∼84% low c-KIT-expressing cells. Using such SGIE containing c-KIT-expressing cells or pure c-KIT-expressing cell fraction isolated from SGIE for implantation will lead most probably to the same result in terms of salivary function.
We further estimated the division ability of cryopreserved SGIE clones, as a tool to assess their potential to reproduce and maintain stem cell features. Our results indicate the same division ability between the nCP and cryopreserved SGIE in all passages examined. This implies that SGIE clones maintain their developmental stage in which they were cryopreserved in, and should not be discriminated from nCP cells in this matter.
Next, an evaluation of cell function by morphological analysis and by protein and mRNA expression was carried out. When grown on Matrigel, recultured SGIE could organize in net-like structures, comprised of acinar-like and ductal-like cell assembly. Moreover, SGIE expressed both proteins and mRNAs related to SG progenitor cells characteristics and to SG functionality. As for the morphological organization, mRNA expression and protein expression were similar for both SGIE mixtures and for SGIE cryopreserved single-cell clones. Consequently, we can comprehend that cryopreservation process does not impair cell function. These findings are in agreement with those previously reported for liver 29 and erythroblast stem cells.
Interestingly, our findings that cryopreservation and re-culture procedures did not alter CK17 expression patterns strengthened the observation of no tumorigenic development in the soft agar test. CK17 is a protein normally expressed in the basal cells of complex epithelia but not in stratified or simple epithelia. 30 CK17 serves also as a marker for basal cell differentiation in complex epithelia. Moreover, CK17 has been found to be overexpressed in malignant head and neck tissues including the tongue, laryngeal cancer, and esophageal squamous cell carcinoma and other tissue including lung and cervical cancers. 31
Transplantation procedures and limitations in the length of the cryopreservation period have been reported as major setbacks in the practice of human hepatocyte transplantation, 32 whereas these obstacles were not encountered using animal hepatocyte models. 29 It has been reported, for instance, that prolonged cryopreservation of human hepatocytes reduces ATP levels due to inhibition of Na+K+-ATPase 33 and results in overall changes in mitochondrial activity. 34 Therefore, although our animal salivary model shows promising results, confirmatory data are required in humans. Our preliminary studies with cultured human SGIE are encouraging (data not shown), but further studies and technological fine-tuning are needed before its clinical implementation.
The results of this study show that a stem cell bank, deriving from an individual's SG, is an achievable goal.SG progenitor cells can be cryopreserved for prolonged periods with little effect on their characteristics following their isolation as single cell-originated clones using a novel, straightforward method. These cells hold promise as candidates for future safe autologous transplantation in humans (Fig. 4).
Footnotes
Acknowledgments
This research was supported in part by the Israel Science Foundation (no. 1083/11) and by the German Israeli Science Foundation (no. 1911–58.2/2006).
Disclosure Statement
The authors indicate no potential conflicts of interest.