
Abstract
Vascular invasion and admixture of the nude mouse cells with seeded cells make it difficult to reapply the regenerated tissues to the restoration of host tissue defects. Therefore, a device that is capable of allowing for autologous or allogenic tissue growth while preventing host tissue invasion will be a valuable tool for in vivo tissue engineering. We have previously fabricated a novel silicon-perforated chamber. The aim of this study was to evaluate whether this chamber, after being implanted subcutaneously in experimental animals, would hinder host tissue ingrowth while providing an environment inside its cavity for in vivo growth of either autologous or allogenic implant cells. We found that the chamber did not induce severe foreign body reaction, and the chambers with perforated pores of 1–3 mm in diameter effectively inhibited the host granulation tissue or vascular invasion for as long as 3 months. In addition, the exudates rich in vascular endothelial growth factor, basic fibroblast growth factor, transforming growth factor-β, insulin-like growth factor-1, and platelet-derived growth factor-BB were steadily generated and collected in the chambers. In vitro cell culture studies revealed that the exudates were able to support the viability and proliferation of rabbit chondrocytes, rat mesenchymal stem cells, and human fibroblasts. The results indicate that this novel chamber could potentially provide an environment favorable for in vivo tissue engineering while effectively preventing host tissue or vascular invasion.
Introduction
Most researchers use immunodeficient animals such as nude mice as animal models in the in vivo regeneration processes of tissue engineering.7–9 Recently, the outcome of conventional tissue engineering products for clinical application is unsatisfactory, leading to the consensus that it would be more promising if the products could be formed in an immunocompetent host.6–8 However, vascular invasion and admixture of the host cells with seeded cells are the critical issues that need to be resolved before in vivo regenerated tissues can be used to repair host tissue defects. Therefore, it is desired to have an environment that would mimic the in vivo regeneration conditions while being isolated from host tissue or vascular invasion.
A model consisting of an atriovenous blood vessel loop inside a semi-sealed polycarbonate chamber has been used in immunocompetent animals such as rats in some tissue-engineering researches.10–12 However, host tissues with the vascular pedicle grew in the chamber leading to secondary deformity of implanted donor tissues. In addition, the host cell ingrowth and vascular invasion facilitated degradation of donor tissues that were eventually replaced by connective tissues. 13 If the cell-seeded scaffold was implanted subcutaneously, the inflammatory reactions in the immunocompetent animals could lead to damage or deformity of the regenerated tissue, which might obstruct the reconstruction, 4 and even the autologous seeded cells would ignite the same problematic events. 14 Furthermore, the host vascular invasion and interference would impair the quality of the regenerated tissue.
In view of the above concerns, we designed a hollow perforated silicone chamber that is suitable for implantation in experimental animals. This chamber has many advantages for tissue-engineering purposes: (1) the firm hollow chamber provides a large isolated space for regenerated tissue; (2) the chamber is easily implanted, located, and retrieved; (3) the size and shape of the chamber can be adjusted according to research requirements; and (4) the holes on the chamber allow the injection of growth factors or differentiation agents for specific tissue regeneration. Our previous research suggested that autologous tissue-engineering cartilage of rabbit could be successfully constructed in this chamber. 15
In present study, we examined the ability of this novel silicon-perforated chamber to support the survival and growth of diverse cells when implanted subcutaneously in experimental animals. We also characterized the dynamic changes of exudates in the chamber. It is revealed that this novel chamber provided an ideal microenvironment where exudates with nutrients and growth factors were dynamically accumulated and sufficiently supported the survival and proliferation of in vivo implanted autologous, allogenic, and heterologous cells. Furthermore, the ingrowth and vascular invasion of host tissues into the chamber were not observed during a 3-month period, suggesting that the chambers with such specific design are able to effectively separate implanted cells from host tissue interference.
Materials and Methods
Experimental animals
Newborn New Zealand white rabbits, adult rabbits, and Sprague-Dawley (SD) rats were purchased from Animal Laboratory, Fourth Military Medical University. All animal experimental protocols were approved by the Animal Care and Experiment Committee of the Fourth Military Medical University.
Preparation of perforated bioinert chamber
The perforated bioinert chamber was made of silicone and in oval shape (Fig. 1). The hollow space inside the chamber communicated with outside environment through pores on the silicone wall. To make a chamber, two identical half-chambers, each with evenly distributed pores drilled on its wall, were prepared in a mold and joined together to form a whole piece of chamber. In this study, we designed three types of chambers, Chamber A, B, and C, with different specifications as shown in Table 1 and Figure 1. All chambers were sterilized by autoclave before being implanted subcutaneously in rats and rabbits.
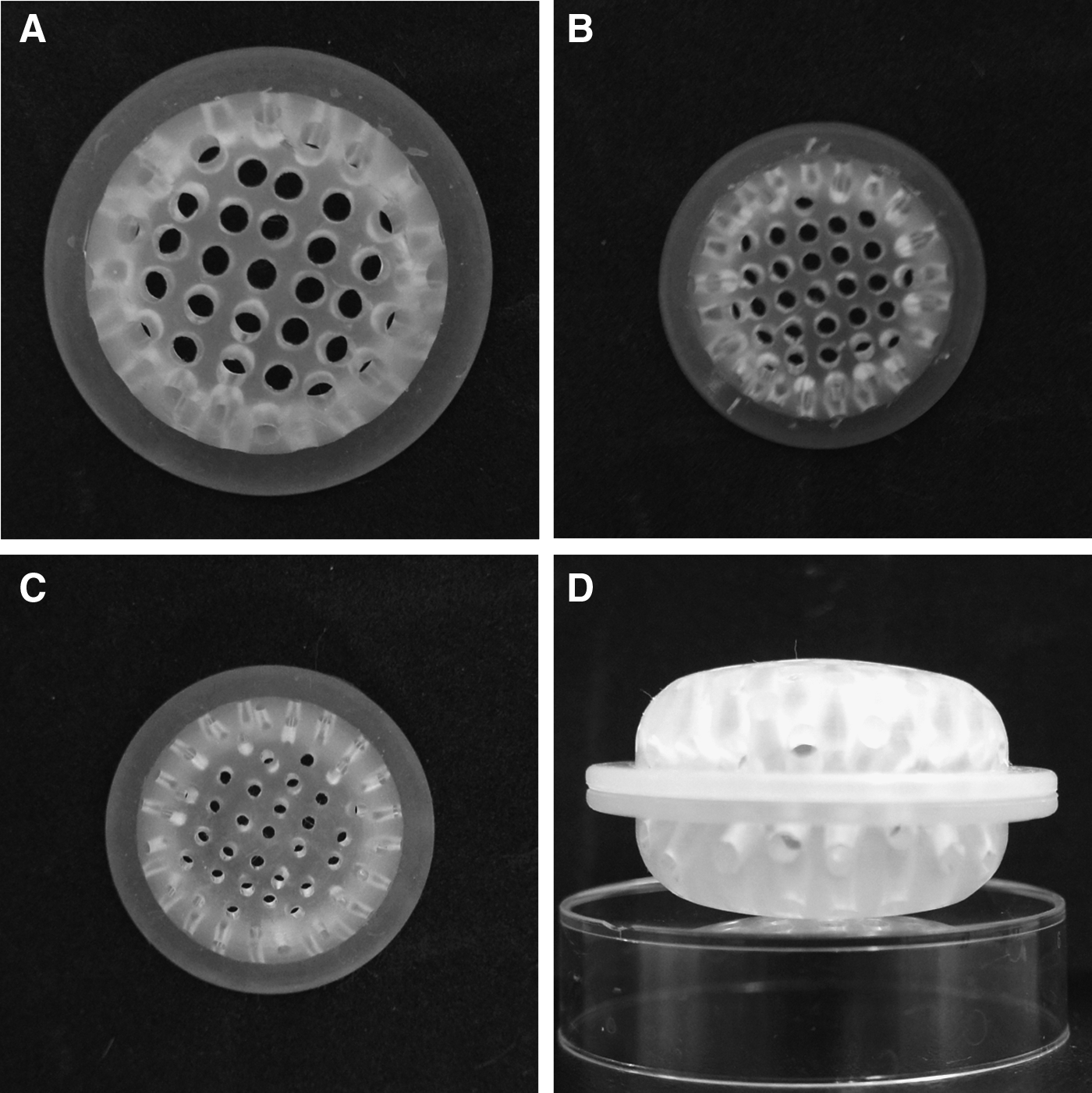
Macroscopic view of the three silicone chambers of different specifications were designed for this study.
Isolation and culture of cells
Rabbit chondrocytes
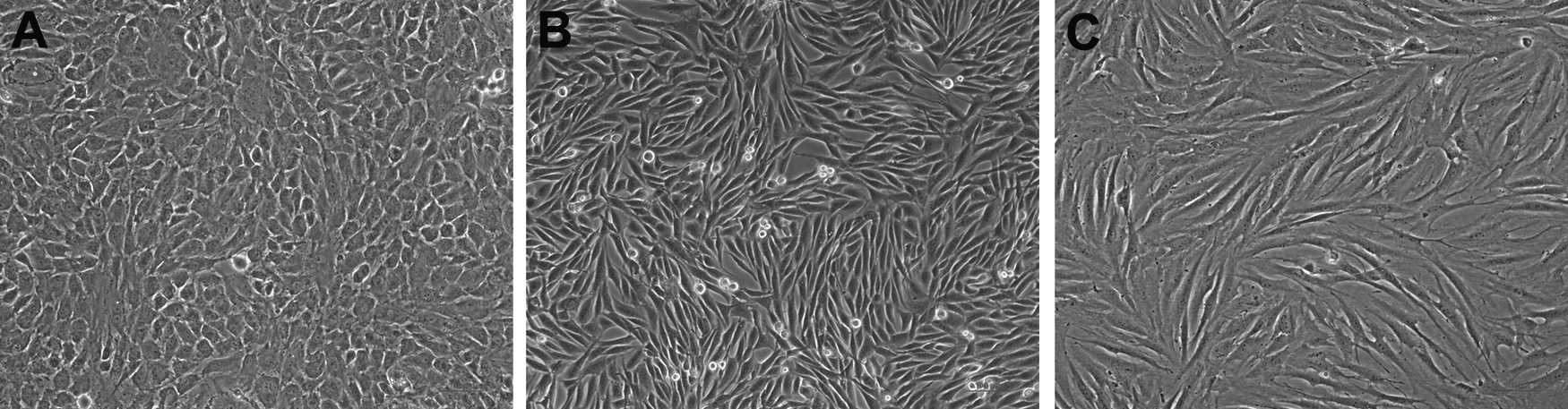
The method for collecting and culturing rabbit chondrocytes was described previously. 16 Briefly, fresh cartilages were collected from the ears of neonatal New Zealand white rabbits and cut into 2×2 mm blocks followed by digestion in 0.25% trypsin/phosphate-buffered saline (PBS) solution with 0.02% ethylenediaminetetraacetic acid (Sigma) at 37°C for 30 min. The tissue blocks were then transferred and digested in 0.1% collagenase II (Sigma) in the serum-free Dulbecco's modified Eagle's medium (DMEM high glucose [HG]; Hyclone) at 37°C for 12–16 h. After pipetting or stirring and passing through a 200-μm filter, the cell suspension liquid was centrifuged and washed for three times with PBS. The resultant chondrocytes were resuspended, counted, and seeded into culture dishes (dia. 10 cm) at a cell density of 2×104/cm2 in DMEM HG with 10% fetal bovine serum (FBS; Hyclone) and 1% penicillin (100 U/mL)–streptomycin (100 μg/mL). The chondrocytes were cultured in a 37°C incubator, and the medium was changed every 3 days. Passage-2 chondrocytes were used for further experiments (Fig. 2).
The appearance of the isolated rabbit chondrocytes
Rat mesenchymal stem cells
We adopted our previously published protocol for the isolation and culture of rat mesenchymal stem cells (MSCs) in this study. 17 Briefly, SD rats weighing from 100 to 120 g were sacrificed by carbon dioxide inhalation and placed in 75% alcohol for 15 min. The femurs and tibias of the SD rats were harvested surgically in a laminar flow hood. The bones were cut open at both ends, and the marrow was flushed with 5 mL of DMEM/Ham's F-12 (DMEM/F-12; Hyclone) through a 22-gauge blunt-end needle. Single-cell suspension was layered over an equal volume of Histopaque1.077 (Sigma) in a 15-mL centrifuge tube. Centrifugation was performed at 2000 rpm for 30 min at room temperature. The mononuclear cells in the interface layer were collected, washed with PBS, and pelleted at 2000 rpm for 5 min. Cells were resuspended in DMEM HG/F12/10% FBS/1% penicillin–streptomycin, and the cell density was adjusted to 3×105/mL. Two milliliters of cell suspension was seeded in a 25-cm2 plastic cell culture flask and cultured in a 37°C incubator. After a series of passages, the attached marrow cells became homogeneous18,19 (Fig. 2).
Human fibroblasts
The fibroblasts were derived from foreskin circumcision specimens of healthy young children (non-neonatal, aged between 5 and 7 years). The protocol of using human foreskin in this study was approved by the Technology Human Research Ethics Committee of the Fourth Military Medical University. Primary fibroblasts were isolated from the skin by mincing the derma into small pieces, and incubated in type I collagenase (0.05%; Sigma) and trypsin (0.02%; Sigma) at 37°C for 6 h. The slurry was then centrifuged at 1200 rpm for 8 min and resuspended in DMEM HG/10% FBS. These cells were then seeded into 25-cm2 plastic cell culture flasks at a density of 3×104/mL. Passage-4 fibroblasts were used for further experiments 20 (Fig. 2).
All the cell cultures were incubated at 37°C in a 5% carbon dioxide/95% air atmosphere.
The implantation of the chambers
Adult rabbits and SD rats were used in the study. After anesthesia by intravenous injection of pentobarbital sodium (1%, 4 mL/kg), the animal's dorsal skin was dissected. The silicone-perforated chamber was then inserted subcutaneously. The time of chamber implantation was set as a time point of day 0. For the transplantation of the Chamber A, two chambers were inserted in the dorsal area of each rabbit (Fig. 3). The Chambers B and C were implanted on the backs of SD rats (Fig. 3), with one chamber per rat. The skin wounds were closed by suture. All surgical procedures were performed under sterile conditions. All the animals were given intensive care and raised in separate cages, and 20 mg/kg ampicillin was given intramuscularly daily for 3 days after the surgery.
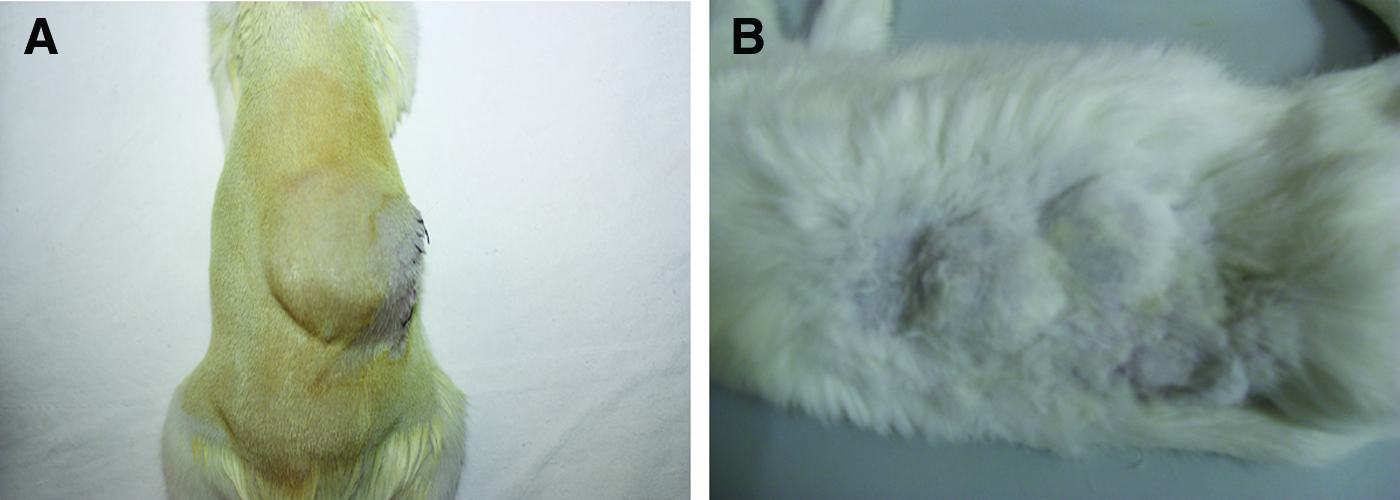
The subcutaneous implantation of the chambers on the backs of the rat
Exudate volume measurement and collection
Ten-milliliter syringe with 26G needle was used to extract the exudate from the chamber, and after the volume was recorded, the exudate was injected back to the chamber. The volumes of exudates measurement in different chambers were recorded daily from the first day after the surgery until the chambers were fulfilled, to determine the effects of shape and size of the chambers on exudate secretion among the three different groups (n=8 in each group).
To collect sufficient exudates for further studies, Type B chambers were implanted in extra-SD rats (n=8) as described previously. The exudates were collected every 2 days from day 3 for 2 weeks. The exudates were centrifuged at 3000 rpm for 5 min, and the supernatants were collected and transferred into several 2-mL microcentrifuge tubes for storage in a −80°C freezer.
In vitro and in vivo cell culture
Cell suspensions of rabbit chondrocytes, rat MSCs, and human fibroblasts at a density of 3×105/mL were seeded in nine 6-well plates, 2 mL/well, respectively. All the wells were added with 2 mL DMEM HG/10% FBS, and the plates were incubated in a 37°C incubator for 24 h to allow the cells to adhere to the wells. After 24 h of incubation, the culture media were removed, and each well was rinsed three times with PBS, and then all the culture wells of the three cell types were supplemented with three media, respectively. For medium Group A, the cells were cultured with 2 mL exudates from the previous collection. For Group B, the cells were cultured with a mixture of 1 mL DMEM HG/10% FBS and 1 mL exudates. For Group C, the cells were cultured with 2 mL DMEM HG/10% FBS only (Fig. 4A). Multiple wells were set up for each culture medium of Group A, B, or C, for each cell type. To investigate whether the exudates could support the long-term survival and growth of the cells from different sources in vitro, all the plates were subsequently cultured for 10 days, and the media were changed every 3 days.

rMSCs, human fibroblasts, and rabbit chondrocytes cultured in vitro with exudates, Dulbecco's modified Eagle's medium (DMEM), and the mixture of DMEM and exudates. Pictures were treated without white balance to distinguish the different culture media (×100).
(3-(4, 5-Dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) assay was used to assess the effects of exudates on cell viability and growth. Human fibroblasts at a density of 3×104/mL were seeded on 96-well culture plates, 150 μL/well, and incubated at 37°C overnight. After all the cells adhered to the wells, the culture media were removed, and each well was washed three times with PBS, and then each well was added with 150 μL of any one of the medium Groups A, B or C, respectively. Six wells were set up for each medium group treatment. All the plates were incubated at 37°C for 10 days, and culture media were replenished every 3 days. At different time points, the cells were given 20 μL of 2 mg/mL MTT solution and incubated at 37°C for 4 h. The medium was then removed, and the resulting intracellular formazan crystal was solubilized in 100 μL/well of dimethylsulfoxide. The optical density at 490 nm, with the reference at 620 nm, was determined using an enzyme-linked immunosorbent assay (ELISA) reader. 21 The absorbances of the media without cells were set as blank control.
To evaluate the viability and growth of rat MSCs inside the subcutaneously transplanted chambers, we first labeled these cells with (3H-Indolium,5-[[[4-(chloromethyl)benzoyl]amino]methyl]-2-[3-(1,3-dihydro-3,3-dimethyl-1-octadecyl-2H-indol-2-ylidene)-1-propenyl]-3,3-dimethyl-1-octadecyl-, chloride (CM-DiI; Molecular Probes, Inc.) following the protocols described previously. 17 Briefly, the freshly isolated rat MSCs were washed and resuspended with PBS and incubated with CM-DiI at a concentration of 2.5 μg/mL PBS at 37°C for 5 min followed by further incubation at 4°C for 15 min. After washing twice with PBS, the cells were mixed into 7 mg/mL collagen I solution (neutralized to pH 7.2–7.4) at a density of 5×107/mL. Hydrogels were formed after incubation at 37°C for 10 min. Then, the hydrogel blocks were transferred into Type C chambers and implanted in the SD rats (n=6) as previously described. Ten days later, the animals were sacrificed by intraperitoneal of overdose pentobarbital sodium. The hydrogel blocks were retrieved, washed with PBS for three times, and digested with 1% collagenase I (Sigma) at 37°C for 2 h to obtain the seeded rat MSCs. 22 The cells were then transferred into culture dishes with a fresh medium (10% FBS) and incubated at 37°C for 24 h to allow the living cells to adhere to the dishes. After the tiny fragments and dead cells were removed by three-time PBS washing, the survival CM-DiI-labeled rat MSCs were inspected with fluorescence microscopy.
Exudates components analysis
Type B chambers were implanted subcutaneously in SD rats (n=8). On days 3, 5, 7, 14, 21, and 30, the exudates were obtained and stored as previously described. An aliquot of each sample as well as normal SD rat serum control and the DMEM HG medium control were evaluated for the levels of total protein (TP), albumin (ALB), glucose (GLU), K+, Na+, and Cl− by a Roche cobas c 311 Autobiochemical Analyzer (Roche), respectively. The levels of vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF), transforming growth factor-β (TGF-β), insulin-like growth factor-1 (IGF-1), and platelet-derived growth factor-BB (PDGF-BB) were also determined, respectively, by ELISA according to the manufacturer's instructions (Bender MedSystems, Inc.). Each sample was measured in triplicate.
Statistical analysis
All results were expressed as mean±standard deviation. SPSS software version 12.0 (SPSS, Inc.) was used to evaluate the statistical significance by analysis of variance followed by SNK post hoc test. A value of p<0.05 was considered statistically significant.
Results
Exudate volume measurement
The appearance of exudates in the chamber was transparent and in light yellow color. The exudate volumes of all the three chambers increased steadily post-transplantation until the chamber cavities were fulfilled (Fig. 5). The fastest exudate volume increase was observed in Type A chamber (1.55±0.21 mL/day), and it took about 8 days for the exudate to fill up the chamber. The rate of exudate volume increase in Type B chamber was about 0.91±0.13 mL/day, and 4 days were needed for the exudates to fill up the chamber. The volume increase in Type C chamber was about 0.67±0.07 mL/day, and it took 6 days for the exudates to fill up the chamber. The differences between the three groups were obvious (p<0.05) (Fig. 5).

The exudate volume-growing curve of the three different chambers. Color images available online at
Cell culture in vitro
Rabbit chondrocytes, rat MSCs, and human fibroblasts were in good growth condition with normal morphology when cultured in DMEM HG/10% FBS (Fig. 2). Twenty-four hours after the culture with exudates or their mixture with DMEM HG/10% FBS, the cells anchored well, proliferated normally, and reached a confluence of 80%–90% at about day 4. (Fig. 4). After passaging by trypsinization with 0.25% trypsin and reseeding at lower cell density (at the ratio of 1:3), all types of cells were incubated continuously in culture media of Groups A, B or C, respectively. The subcultured cells were again able to reach 100% confluence. The data suggest that the cells from different sources not only survive but grow well in the exudates or the culture media containing the exudates.
To confirm the effects of exudates on the survival and proliferation of cells, we cultured human fibroblasts in 100% exudates (Group A) or in DMEM HG/10% FBS containing 50% exudates (Group B). The fibroblasts cultured in DMEM HG/10% FBS was also included as control (Group C). The MTT assay was used to assess the cell growth under these different conditions. As shown in Figure 6, fibroblasts cultured in either Group A or Group B grew significantly faster than Group C. At day 6, Groups A and B medium-treated fibroblasts reached almost 100% confluence. In comparison, fibroblasts in Group C only reached about 70% confluence in the 96-well culture plates and continued to proliferate until day 10 when the 100% confluence was achieved. The growth rates were significant different between Group A versus C and between Group B versus C at day 6 (p<0.05), whereas no significant differences of growth were observed between Group A and B medium-treated cells over the 10-day culture period.
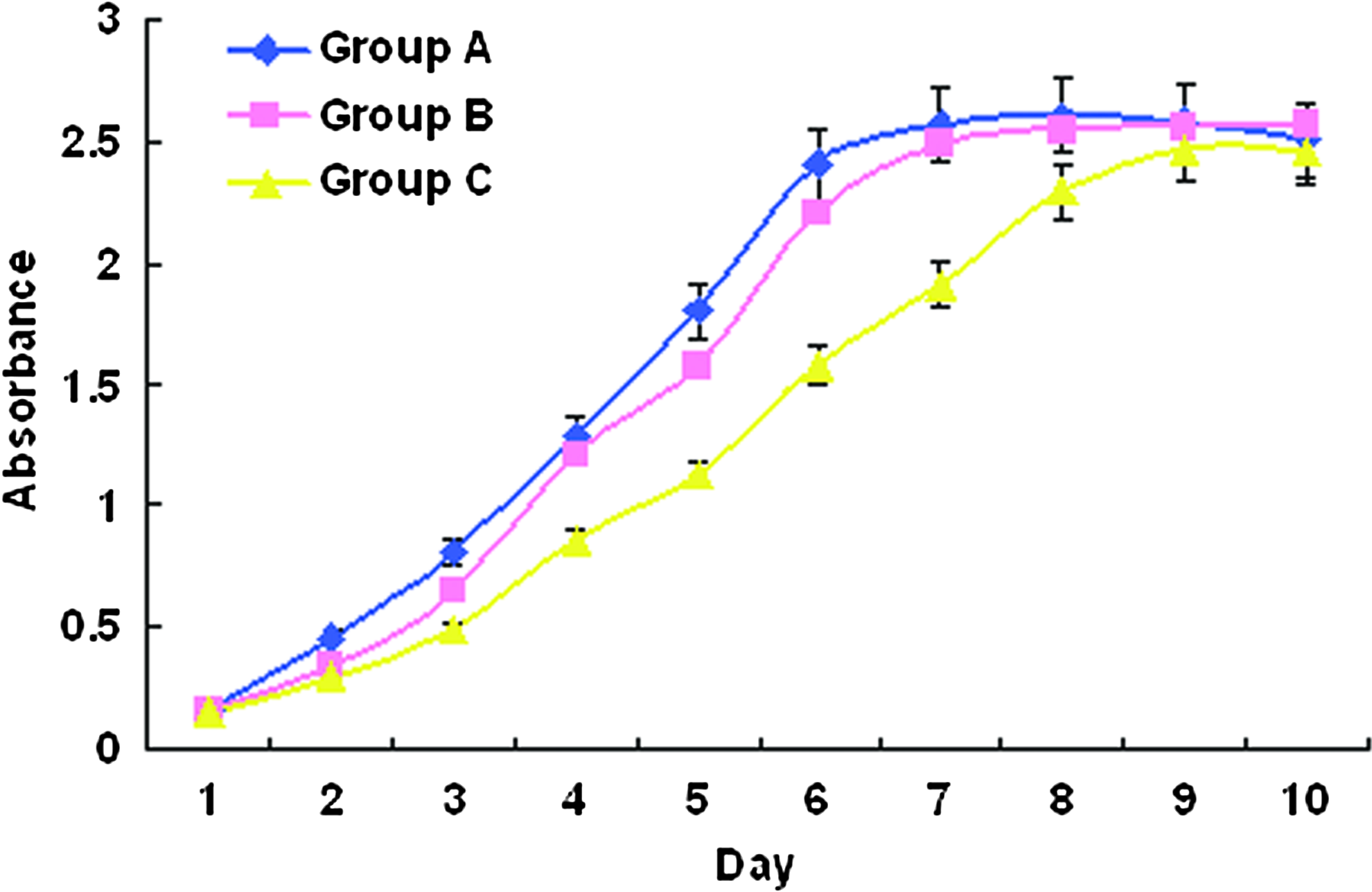
The curve of MTT assay showing the effects of exudates (Group A), DMEM (Group C), and the mixture of DMEM and exudates (Group B) on human fibroblast viability and growth. MTT, (3-(4, 5-Dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide Color images available online at
Cell culture in vivo
To evaluate whether the implanted chambers provide an environment suitable for the maintenance of cell viability, we first inserted the collagen I hydrogels containing CM-DiI-labeled rat MSCs in Chambers of type C, followed by implantation subcutaneously in the rats. Ten days later, when incisions were made on the dorsal skin of rats and the chambers were opened, vast and bright exudates were observed. All the hydrogel blocks were immersed in the exudate fluid. Interestingly, no apparent host tissue ingrowth and vascular invasion were observed in all the chambers in our experiments. In addition, when the cells were retrieved from hydrogels by collagenase I digestion, and transferred into culture dishes for a 24-h culture in DMEM HG/10% FBS, CM-DiI-labeled cells attached and spread well to the dishes and showed a typical morphology of rat MSCs (Fig. 7). Therefore, these results suggested that the exudates and microenvironment in the chamber could support the survival of implanted cells in vivo. Moreover, these results also indicated that the chambers possess certain characteristics unfavorable for host tissue ingrowth and vascular invasion over a period of time (up to 3 months, data not shown).

The CM-DiI-stained rMSCs cultured in vivo in the tissue-engineering chamber in rat. (×100). The isolated and CM-DiI-stained rMSCs, before implanting into rat, under white light
Exudates components analysis
The levels of TP, ALB, GLU, K+, Na+, and Cl− of either the exudate samples or the serum samples were consistent over 30 days after chamber transplantations (Fig. 8A, B). However, the exudate levels of TP and K+ were significantly lower than that of the corresponding serum samples (p<0.05) (Fig. 9), whereas the levels of ALB, GLU, Na+ and Cl− were comparable between serum and exudate samples (p>0.05). Furthermore, in comparison with the normal culture medium (DMEM HG, 10% FBS), the exudate levels of TP and ALB were significantly higher (p<0.05), while the levels of GLU, K+, Na+, and Cl− were markedly lower (p<0.05) (Fig. 9).

Components analysis of exudates on the 3rd, 5th, 7th, 14th, 21st, and 30th day after the implantation of the chambers.

Components comparisons between the exudates, normal rat serum, and DMEM.
The levels of VEGF, bFGF, TGF-β, IGF-1, and PDGF-BB maintained stable in all the exudates collected over a 30-day/month period (Fig. 10). Compared with the serum and normal culture medium, the concentrations of the growth factors in the exudates remained in the level (ng/mL or pg/mL of each factor, mean±standard deviation) that were significantly lower than the serum (p<0.05), but higher than the normal culture medium used in the experiments (p<0.05) (Fig. 11).
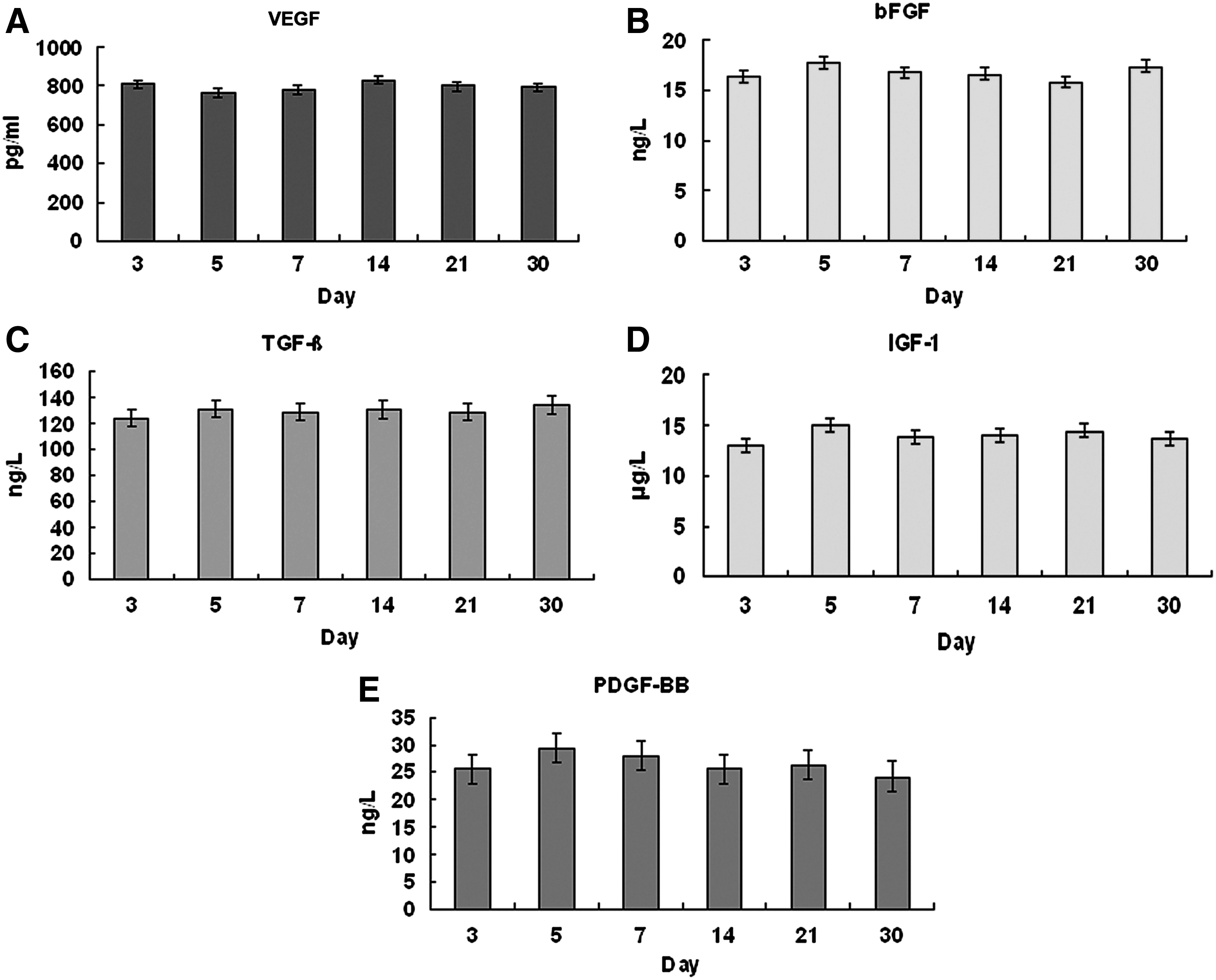
The levels of vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF), transforming growth factor-β (TGF-β), insulin-like growth factor-1 (IGF-1), and platelet-derived growth factor-BB (PDGF-BB) in the exudates on the 3rd, 5th, 7th, 14th, 21st, and 30th day after the implantation of the chambers.
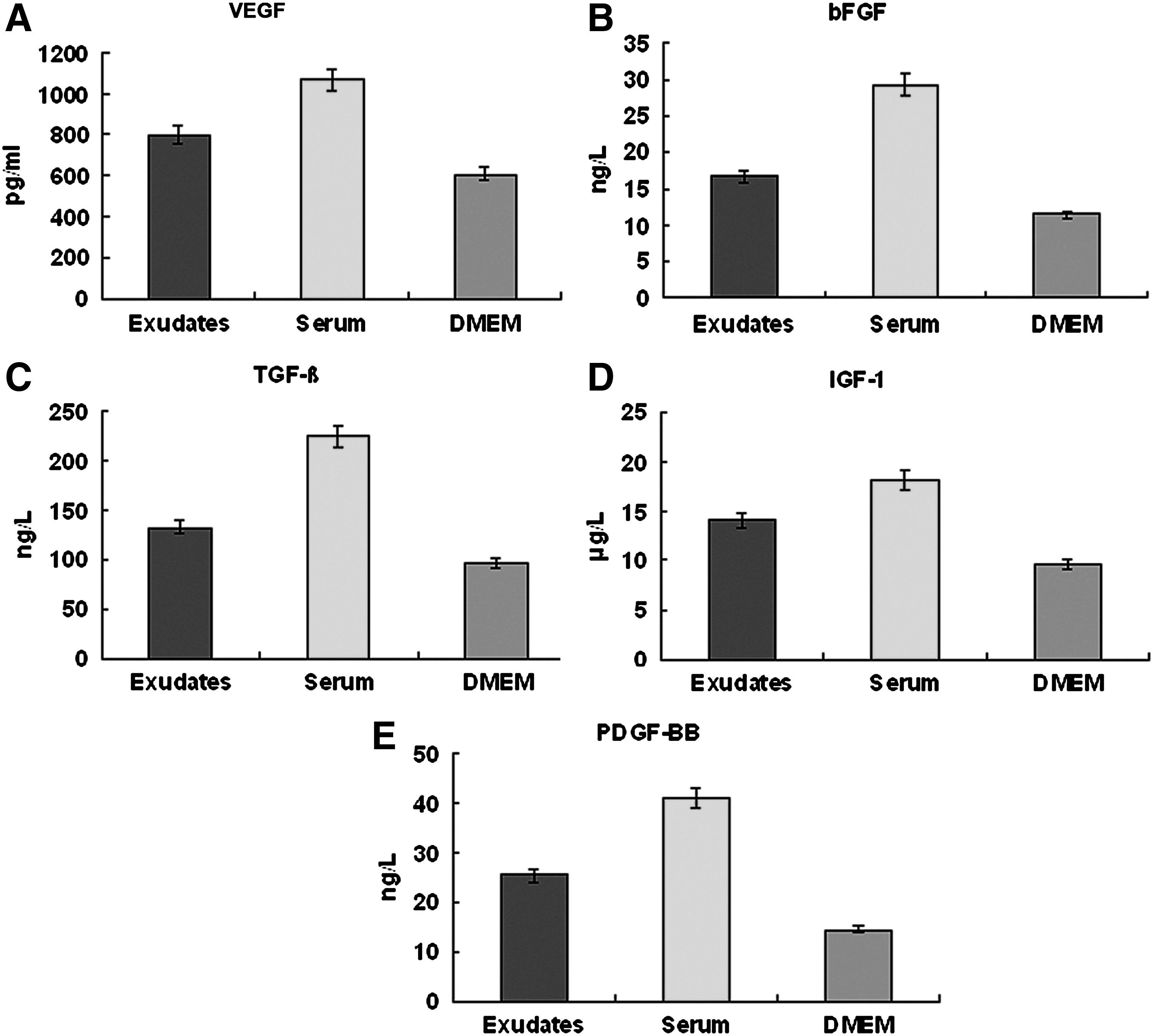
The levels of VEGF, bFGF, TGF-β, IGF-1, and PDGF-BB in the exudates, normal rat serum, and the DMEM.
Discussion
The microenvironment of tissue engineering chamber had not been previously examined in detail. Little attempt has been made to characterize whether this model could provide an ideal space for cell survival and growth in vivo. Tremendous progress has been made in the design of devices that significantly promote the development of experimental tissue engineering10–12,23–25; however, these device models did not supply an enclosed environment; therefore, host cell ingrowth and vascular invasion were inevitable that result in the damage or deformity of the regenerated tissue. In the present study, we designed and used silicon-perforated chambers that were suitable for in vivo transplantation. Our results proved that these chambers provided an environment that allowed for the transmission of body fluid and nutrition into the chambers and dramatically inhibited the host cell ingrowth and vascular invasion into the chamber cavity.
Silicone, a bioinert material, has been widely used in medical field for many years, and its good biocompatibility has been recognized. The chambers used in this study did not cause significant foreign body reaction after implanted subcutaneously into the animals, and it could provide a large isolated steady space for the regenerated tissue. The sphere design allowed the maximal contact of chambers with the surrounding tissues, consequently, promoted a fast exudate production inside the chambers. The exudate volume measurement showed that the tissue fluid infiltration rate varied according to the specifications of the chambers. The lager the holes on the wall of chamber, the faster the exudates generated, indicating that the diameter of the holes was the key factor that influenced the infiltration speed (Fig. 6).
Most cells proliferate steadily when cultured in optimal conditions defined by varied nutrients, appropriate temperature, osmotic pressure, atmosphere, pH, nontoxic, and sterile environment, etc.26–28 In terms of autologous and allogeneic engineering tissue regeneration in vivo, it is generally believed that an abundant and rapid blood supply is needed to meet the requirement of the optimal conditions.10,12,29,30 However, host tissue invasion that leads to the disintegration and difficulty of retrieval of engineered tissues still remains a great challenge in these in vivo models. We previously reported that an implantable perforated chamber effectively blocked host tissue invasion and provided an isolated space for in vivo cultivation of chondrocyte/collagen implants. 15 Since accumulated exudates were collected inside the chambers, we speculated that sufficient nutrients were also provided herein to support the survival and growth of chondrocytes. Indeed, in our study, we showed that the rabbit chondrocytes, rat MSCs, and human fibroblasts were able to survive and grow when cultured in the exudates. These results suggest that the exudates contained sufficient nutrients to keep cell survival in vitro. Besides, the healthy and CM-DiI-positive cells in the hydrogel blocks could be retrieved and cultured continuously in vitro after grown in implanted chambers, indicating that the exudates in the chamber did provide an environment for in vivo cell growth and regeneration of engineered tissues.
Physiologically, most cells of multicellular organs rely on the interstitial for constant nutrient supply, and interstitial fluid is actually the infiltrate of plasma. 31 Upon the surgical insertion of the perforated chambers, trauma and silicone material will immediately initiate host tissue responses resulting in the accumulation of inflammatory exudates in the chambers. Based on our analysis, the exudates are similar, with slightly lower TP and K+, in composition to serum samples derived from the experimental animals. Further analysis also revealed that the chamber exudates contained comparable, although lower, levels of VEGF, bFGF, TGF-β, IGF-1, and PDGF-BB. Besides, the concentrations of the growth factors in exudates were significantly higher than those in the culture media, suggesting that multiple intracellular pathways would be activated and facilitated cell survival and proliferation. Indeed, human fibroblasts grew faster and retained higher viability in 100% and DMEM HG/50% exudates than in a normal culture medium that was comprised of DMEM HG/10% FBS (Fig. 7). These results show that the exudates are capable of initiating cell proliferation required by tissue regeneration processes. 32
It is noted that growth factors play important roles in the tissue engineering research. The five growth factors we tested have been widely studied in tissue regeneration models.33,34 VEGF is a cell-produced signal protein that has multiple biological attributes besides stimulating vasculogenesis and angiogenesis. 34 VEGF is believed to promote differentiation of stem cells into endothelial cells 35 and boost the function of MSCs and endothelial progenitor cells. 36 bFGF has been referred to as pluripotent growth factor due to its multiple actions on multiple cell types. 37 TGF-β controls proliferation, cellular differentiation, and other functions in most cells. In particular, it is critically involved in cartilage tissue engineering.38,39 IGF-1 promotes cell growth and induces regeneration of muscle and cartilage.40,41 PDGF-BB regulates cell growth and division, and is a potent mitogen for cells of mesenchymal origin. Reports have suggested PDGF-BB could stimulate mesenchymal cell proliferation, migration, and preadipocyte differentiation and adipogenesis.11,42,43 Therefore, we believe that the chamber exudates containing these growth factors provide an ideal environment for in vivo tissue engineering, although the specific effects of each growth factor need further investigation.
Conclusion
We have established a novel model using the silicon-perforated chamber for in vivo tissue engineering research. This new transplantable chamber not only provides a stable environment that can achieve transmission of body fluid, but also prevents host cell ingrowth and vascular invasion. In addition, the proproliferation effects of exudates imply of a potential application of the chambers in the field of tissue engineering.
Footnotes
Acknowledgments
The authors thank Professor Xunzhen Zhou, Department of Foreign Language, for assisting in the preparation, and Dr. Yingjun Su, Department of Plastic Surgery Center, Xijing Hospital, Xi'an, for critical reading of this manuscript.
Disclosure Statement
No competing financial interests exist.