
Abstract
Biomaterial-based tissue-engineered tumor models are now widely used in cancer biology studies. However, specific methods for efficient and reliable cell seeding into these and tissue-engineering constructs used for regenerative medicine often remain poorly defined. Here, we describe a capillary force-based method for seeding the human prostate cancer cell lines M12 and LNCaP C4-2 into sphere-templated poly(2-hydroxyethyl methacrylate) hydrogels. The capillary force seeding method improved the cell number and distribution within the porous scaffolds compared to well-established protocols such as static and centrifugation seeding. Seeding efficiency was found to be strongly dependent on the rounded cell diameter relative to the pore diameter and pore interconnect size, parameters that can be controllably modulated during scaffold fabrication. Cell seeding efficiency was evaluated quantitatively using a PicoGreen DNA assay, which demonstrated some variation in cell retention using the capillary force method. When cultured within the porous hydrogels, both cell lines attached and proliferated within the network, but histology showed the formation of a necrotic zone by 7 days likely due to oxygen and nutrient diffusional limitations. The necrotic zone thickness was decreased by dynamically culturing cells in an orbital shaker. Proliferation analysis showed that despite a variable seeding efficiency, by 7 days in culture, scaffolds contained a roughly consistent number of cells as they proliferated to fill the pores of the scaffold. These studies demonstrate that sphere-templated polymeric scaffolds have the potential to serve as an adaptable cell culture substrate for engineering a three-dimensional prostate cancer model.
Introduction
One of the main obstacles in generating these models or other tissue-engineered constructs is developing efficient and reliable methods to seed cells within scaffolds. A variety of seeding protocols have been described in the literature, including simple static seeding, 13 spinner flasks, 14 perfusion bioreactors, 15 injection, 16 centrifugation, 17 and vacuum filtration, 18 among others. However, in many articles, details regarding this critical step in the tissue-engineering process remain vague or unmentioned.
Seeding optimization is strongly dependent on scaffold architecture. Biomaterials fabricated by sphere-templating have been used for cardiac tissue engineering, 19 bone regeneration, 20 percutaneous devices, 21 and fundamental studies of the foreign body response. 22 Sphere-templated materials are composed of a network of interconnected spherical pores of uniform size displaying an inverted colloidal crystal geometry (see Fig. 1). This architecture can create difficulties for cell seeding because small pore sizes and pore interconnect diameters restrict the cells from infiltrating the scaffolds. However, the sphere-templating fabrication protocol allows for controllable modulation of these parameters, and thus, the empirical optimization of cell seeding. Here, we describe a quick, simple, and inexpensive method using capillary force to seed two prostate cancer cell lines into sphere-templated porous nondegradable crosslinked poly(2-hydroxyethyl methacrylate) (pHEMA) hydrogels. Other challenges for in vitro tissue engineering, including the optimization of cell culture conditions and the analysis of 3D cell proliferation within a nondegradable construct, are also addressed. In future studies, these techniques can be used to tissue engineer prostate cancer xenografts with a defined local microenvironment.

Sphere-templated pHEMA scaffold morphology.
Materials and Methods
Scaffold fabrication
Sphere-templated pHEMA scaffolds were prepared as previously described. 22 Briefly, spherical poly(methyl methacrylate) beads (Kupa, Inc.) were sieved using an ATM sonic sifter to a uniform size range of 74–86 μm. The beads were inserted into 1-mm-thick glass rectangular molds and sonicated for 20 min to ensure particle packing. The molds were then sintered overnight at 140°C for 22 or 24 h in a convection oven (Thermo Scientific) to fuse the beads at points of contact. pHEMA was prepared by first infiltrating the sintered molds with a mixture comprised of monomeric HEMA (Opthalmic grade; Polysciences, Inc.) (60% vol/vol), cross-linker tetraethylene glycol dimethacrylate (TEGDMA, Polysciences, Inc.) (1.5% mol/mol HEMA), initiator 2,2-dimethyoxy-2-phenylacetophenone (IRGACURE 651; Ciba) (1% mol/mol HEMA), ethylene glycol (JT Baker) (17% vol/vol), water (18% vol/vol), and cell attachment substrate collagen I (BD Biosciences) (15% wt/vol). Polymerization was photoinitiated under a UV lamp (Hanovia) for 10 min. The polymer-bead cakes were then continuously washed with dichloromethane for 72 h in a soxhlet extractor, solubilizing the PMMA beads and leaving a crosslinked polymer network with uniform spherical pores interconnected at the regions where the beads were sintered together. The polymer scaffolds were sterilized for 48 h in 70% ethanol before rehydration in sterile phosphate-buffered saline and cell culture.
Cell culture
After rehydration, pHEMA scaffolds were punched into 6-mm discs using sterile biopsy punches (Acuderm) and preincubated in media for 1 h at 37°C before cell seeding. Two human prostate cancer cell lines, LNCaP C4-2 (from R.A. Sikes, University of Delaware) and M12, 23 were used in this study. LNCaP C4-2 cells were cultured in RPMI 1640 containing 10% fetal bovine serum (FBS). M12 cells were cultured in RMPI supplemented with 5% FBS, 10 μg/mL insulin, 5.5 μg/mL transferrin, 6.7 ng/mL sodium selenite, 250 ng/mL amphotericin B, 50 μg/mL gentamycin, 10 ng/mL epidermal growth factor, and 78 ng/mL dexamethasone (all media and supplements from Cellgro). All 3D cell cultures within scaffolds were performed in 24-well plates containing 1 mL of culture media and a custom sterile stainless steel mesh platform that raised the scaffold ∼1.5 mm and allowed media to circulate under the scaffold. Those plates were placed on an orbital shaker set to 300 rpm within a tissue culture incubator. Culture media were replaced every 2 days.
Cell seeding
Cells were seeded into the scaffolds using three methods. For static and centrifugation seeding, scaffold discs were fit into the wells of a 96-well plate that matched their 6-mm diameter to direct cell infiltration into, and not around, the scaffolds. A 200 μL suspension of 1×106 cells was pipetted on top of the scaffold. For centrifugation seeding, the cell suspensions were centrifuged into the scaffolds at 200 g for 15 min. For capillary force seeding, scaffolds were placed on top of a stack of five to six autoclaved Kimwipes (Kimberly-Clark) and 50 μL of a 1×107 cells/mL suspension was twice layered on top of each scaffold, allowing cells to be drawn into the pores. For all methods, seeded cells were allowed to attach to the scaffolds overnight before being transferred to a 24-well dynamic culture in an orbital shaker.
Histological analysis
Cells were cultured in vitro for up to 7 days within the scaffold. Cell proliferation was tracked qualitatively using histological methods. Scaffolds were fixed overnight at 4°C in a solution of 90% methanol and 10% acetic acid. After fixation, the scaffolds were dehydrated in ethanol and cleared with xylene before paraffin embedding. The embedded scaffolds were sectioned into 5-μm-thick sections with a Leica microtome and placed on positively charged slides (Fisher) that prevented pHEMA section detachment during staining. Fixed paraffin sections were heated at 53°C for 30 min before xylene deparaffinization and rehydration through a graded ethanol series. Slides were stained using a standard hematoxylin and eosin (H&E) protocol and imaged on a Nikon E800 microscope equipped with Metamorph software (version 6.0; Molecular Devices).
Quantitative proliferation analysis
Seeding efficacy and proliferation of M12 and LNCaP C4-2 cells within the scaffolds were quantitatively measured using a PicoGreen® DNA assay (Invitrogen). To establish working sample dilutions and a cell number reference, fluorescence standard curves were produced by freeze–thawing triplicate aliquots of M12 and LNCaP C4-2 cells from 2D culture in 1 mL nuclease-free water at −80°C (Ambion). Samples ranged from 6.25×104 cells to 2×106 cells. To determine the minimum dilution necessary to eliminate PicoGreen fluorescence quenching, standard curve samples were serially diluted from 1:1 to 1:50 in the TE buffer. 100 μL of diluted DNA sample was added to 100 μL of 1× PicoGreen in black-walled 96-well plates and 480 nm excitation/525 nm emission fluorescence was measured using a Safire2 microplate reader (Tecan). 1:20 and 1:50 sample dilutions were sufficient to eliminate quenching concerns for M12 and LNCaP C4-2 cells, respectively.
To determine seeding efficacy and proliferation in three dimensions, scaffolds were seeded with M12 and LNCaP C4-2 cells for 0-, 1-, 3-, 5-, and 7-day proliferation endpoints. Five day 0 (D0) samples for each cell line were frozen immediately after seeding, whereas other samples were cultured in triplicate as described until their respective endpoints. Scaffolds were frozen at −80°C in 1 mL of nuclease-free water. After thawing, 0.1% Triton-X-100 detergent was added and samples were vortexed for 60 s, sonicated for 10 min, and vortexed again before 1:20 (M12) or 1:50 (LNCaP C4-2) sample dilution in the TE buffer for analysis. Cell samples from 2D culture for a same-day standard curve comparison were treated the same and analyzed along with all scaffold samples. 100 μL of a diluted DNA sample was added to 100 μL of 1× PicoGreen and fluorescence was measured. Linear standard curves of 480/525 nm fluorescence versus the cell number were prepared and the 3D scaffold cell content was determined. To confirm effective decellularization of the scaffolds, freeze–thawed scaffolds were fixed, paraffin processed, and stained with H&E as described.
Scanning electron microscopy
Scanning electron microscopy (SEM) was performed at the University of Washington Nanotech User Facility. Scaffolds were frozen, lyophilized, and gold sputter-coated before observation on an FEI Sirion scanning electron microscope.
Digital volumetric imaging
Digital volumetric imaging (DVI) was performed using an apparatus manufactured by The Microscience Group, Inc. After fixation, scaffolds were stained with the fluorescent dyes eosin Y and acridine orange en bloc (whole-mount), dehydrated in a graded ethanol series to xylene, and embedded in opacified epoxy resin. This opacification blocked out-of-plane fluorescence for clearer imaging of the current section. Blocks were serially sectioned with an automated diamond knife microtome. After each section was cut, a fluorescence microscope and digital camera were used to capture a multiwavelength image of the block face, and the block was advanced. Sections were acquired at either 0.9- or 0.45-μm thickness (corresponding to 10× or 20× magnification, respectively), and a typical DVI acquisition consisted of 300 to 1000 sections. The resulting aligned stack of images was compiled into a 3D data set. Custom software allowed data rendering and navigation in all orientations, as well as segmentation of regions of interest. 24
Results
In vitro cell seeding
Figure 1 shows representative DVI and SEM images of sphere-templated pHEMA scaffolds displaying a network of interconnected spherical pores around 80 μm in diameter. Cell seeding into this construct was performed using static, centrifugation, and capillary force-based techniques. Figure 2 shows H&E-stained histological cross sections and DVI of cell-seeded scaffolds. As evaluated qualitatively by histology, the capillary force method was more successful at loading cells within the scaffolds than static seeding or centrifugation, which resulted in fewer cells in the construct and cells largely being confined to surface pore layers. In contrast, the capillary force-seeding technique yielded a higher cell content and a relatively even cell distribution within the scaffolds immediately after seeding (see Fig. 2A, D, F, G). Cell infiltration for the capillary force method was empirically optimized for each cell line by modulating sintering time during scaffold fabrication, which altered the pore interconnect (throat) diameter. Cell navigation through these pore interconnects during seeding is likely a constraint on seeding efficacy, where the cell size relative to the pore throat diameter is a critical parameter. The sphere-templated scaffold has high pore interconnectivity, and many pores have multiple pore throats. As cell suspensions move through the scaffold, smaller pore throats will be occluded by trapped cells, but larger pore throats exist for cells to pass successfully through. As evaluated by SEM, 22-h sintering on 76–84 μm PMMA particles yielded an average pore throat diameter of 19.3±1.7 μm and 24-h sintering yielded an average pore throat diameter of 20.9±2.5 μm. Twenty-two hours sintering was most effective for seeding M12 cells, which have a rounded cell diameter of 11.8±2.0 μm, whereas 24-h sintering worked best for the larger LNCaPC4-2 cells, which have a rounded cell diameter of 17.1±2.6 μm. Although the difference in the pore throat size between 22- and 24-h sintering times was not statistically significant, it was large enough to affect seeding. For the smaller M12 cells, the increase of 1.6 μm/2.4 μm (average/upper bound) for the 24-h sintered scaffolds was enough to allow many more M12 cells to pass entirely through the scaffolds. Conversely, for the LNCaP C4-2 cells, the smaller pores associated with the 22-h sintered scaffolds resulted in more cells getting stuck as they navigated through the porous network.
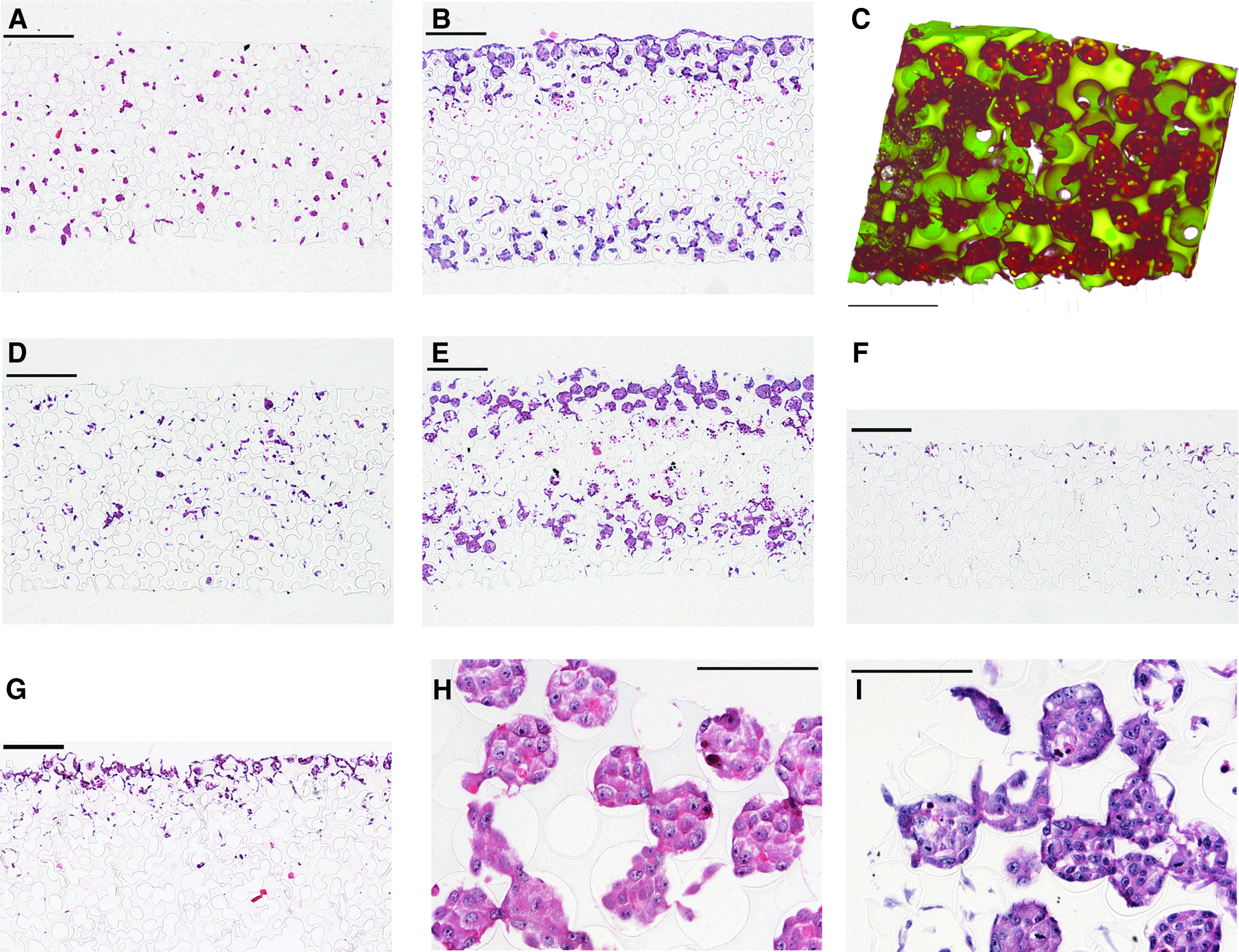
Hematoxylin and eosin-stained histological cross sections and DVI of sphere-templated pHEMA with seeded prostate cancer cells.
In vitro cell culture
Histological images of M12 and LNCaPC4-2 cells seeded within the pHEMA scaffolds showed consistent patterns of growth between the cell lines. After 24 h, cells had attached to the collagen-embedded pore walls of the scaffold, and by 7 days, cells had proliferated to fill many of the pores and the interconnects between the pores (see Fig. 2C, H, I). By 7 days in culture, a necrotic zone was noted in the center of the scaffold (see Fig. 2B, E), presumably due to limitations in nutrient/oxygen diffusion and waste exchange. The distance penetrated into the scaffold by cells at 7 days under static culture was 145.2±29.3 μm, which corresponds approximately to the limit of oxygen diffusion in tissues. The size of the necrotic zone decreased when scaffolds were placed in dynamic culture using an orbital shaker, with convection increasing the distance penetrated by cells to 237.3±33.0 μm. In addition, stainless steel mesh inserts that allowed media to circulate under the scaffolds improved cell viability on the bottom scaffold surface.
Because comparisons between biomaterial scaffold-derived grafts and traditional xenografts in vivo require seeded cell quantification, a PicoGreen DNA assay was developed to track cell seeding efficacy and proliferation within the scaffolds (see Fig. 3). During capillary force seeding, it is possible that some cells will pass all the way through the porous network and, thus, will not be retained after being applied to the scaffold. Indeed, we found variability in the number of cells present after seeding on D0, with seeding efficacies ranging from 27.3%–66.5% for M12 cells and 37.0%–72.8% for LNCaP C4-2 cells. Both cell lines also experienced an average loss of cells by D1, which can be potentially explained by cells on the top surface or bottom pore layers of the scaffold falling off or out of the scaffold when it was transferred to dynamic culture. Both cell lines showed significant increases in cell number on D3, D5, and D7 compared to D1. M12 cell number reached ∼1×106 by D3 and this value remained statistically equivalent over D5 and D7. This consistency may be attributed to cells reaching their proliferation capacity within the pores that are not diffusion limited. LNCaP C4-2 cells showed a more gradual increase in cell number over time, with no significant difference between D3 and D5, but a significant increase from D5 to D7.
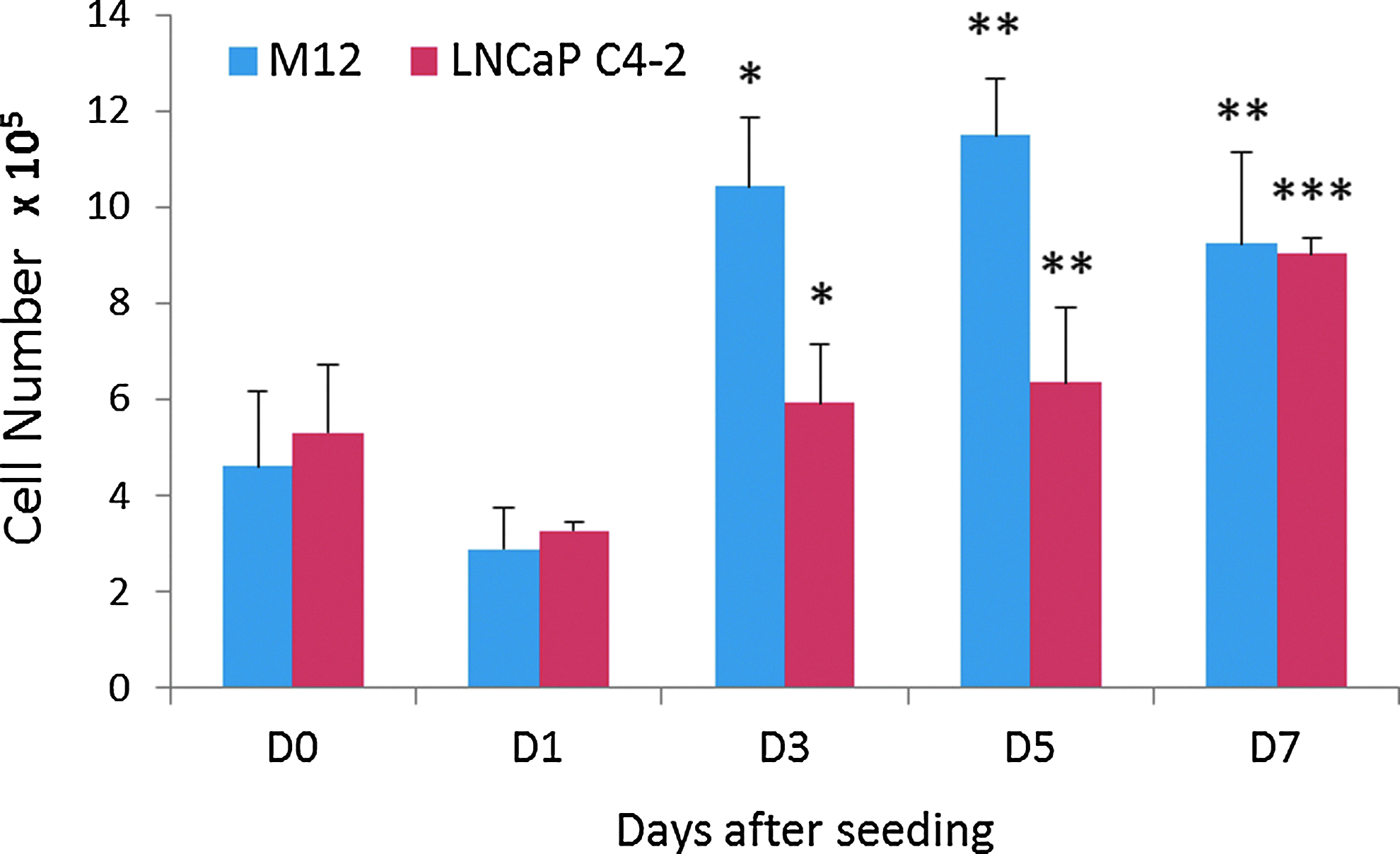
PicoGreen DNA analysis showing M12 and LNCaP C4-2 cell seeding efficiency and proliferation within sphere-templated pHEMA scaffolds sintered for 22- and 24-h, respectively. With 1×106 cells applied by capillary force, both cell lines show average D0 seeding efficiencies around 50%, D1 cell loss, and a significant overall cell proliferation over 7-day culture (*p<0.05 compared to D1, **p<0.01 compared to D1, ***p<0.01 compared to D1 and D5). Color images available online at
Discussion
In these studies, we have developed in vitro cell seeding and culture methodologies that can be used in the generation and analysis of engineered tumor models based on sphere-templated pHEMA scaffolds. Capillary force-based cell seeding of these scaffolds can be empirically optimized by modulating the following parameters: the scaffold pore size, sintering time (interconnect throat size), cell suspension concentration, and the number of times an aliquot of cells is applied to the scaffold. M12 and LNCaP C4-2 prostate cancer cells were most effectively seeded into scaffolds with a pore size around 80 μm using 22- and 24-h sintering times, respectively. A longer sintering time accommodated the larger LNCaP C4-2 cells by allowing them to pass through larger pore interconnects. It should be noted that the same sintering times with different pore sizes (i.e., different template sphere sizes) yielded suboptimal results, so these parameters need to both be adjusted for each cell line. For this reason, optimization for coculture studies where there are large differences in size between cell types could be challenging. In addition, for larger cells, it may be difficult to seed small pore size materials using capillary force since most cells will be confined to the surface pore layers. This pore size limitation is particularly important because it has been demonstrated that a 38-μm pore size is optimal for biointegration upon material implantation, with highest vascularity and lowest fibrous capsule thickness. 22 To potentially avoid seeding limitations for small pores, a thermoresponsive poly(N-isopropylacrylamide) scaffold can be seeded at room temperature, and then cultured at 37°C, as demonstrated by Galperin et al.25,26 The expanded pores at 25°C will shrink at 37°C and trap cells in smaller pores. At the other extreme, pores that are too large may lead to significant cell loss using the capillary force technique since cells will too easily pass through the pore network and leave the scaffold. In this case, the sintering time may be reduced to decrease the pore interconnect size or other seeding methodologies may be more appropriate.
Varying cell suspension concentrations and the number of times a suspension is applied to the scaffold can also impact seeding efficiency. In this case, the application of 1×106 total cells from twice layering 50 μL of a 1×107 cells/mL suspension was optimal. Applying too dilute a suspension many times usually resulted in a significant cell loss, while too concentrated a suspension yielded cells clustered in one or two surface pore layers. Material selection also makes a difference; if cells adhere strongly to the material, they are less likely to travel quickly through it and seeding efficiency may be improved. These studies utilized pHEMA with embedded collagen I. Without the collagen, the low cell adhesion associated with the hydrophilic pHEMA may have reduced the seeding efficacy. Finally, it should be noted that cancer cells may be more amenable to this type of seeding than other nontransformed cell types due to their enhanced elasticity, 27 which may have provided them the capacity to deform while navigating the porous scaffold network. That said, capillary force seeding subjects cells to a minimal amount of force compared with methods such as vacuum filtration, which can shear and deform cells.
Once seeded, maintaining viable cells within a sphere-templated scaffold in vitro for longer than a few days also presented challenges. It was determined that necrotic zone thickness could be reduced using the convection provided by a simple orbital shaker. This enhanced cell penetration into the scaffold using dynamic culture agrees with previously demonstrated results with inverted colloidal crystal scaffolds. 28 In addition, scaffolds in both static and dynamic culture lost viable cells over 7 days along the edge of the scaffold lying on the bottom of the tissue culture plate. To compensate for this, a stainless steel mesh platform was used to raise the scaffold and allow media to flow under the scaffold, which significantly improved cell viability and decreased the necrotic zone size.
Concerns regarding the measurement of cell proliferation in three dimensions have been expressed. 29 In a nondegradable tortuous construct, extracting viable cells is a difficult if not impossible task. Non-endpoint metabolic analyses such as alamarBlue® are limited by diffusional concerns, and it was observed that proliferation trends measured by alamarBlue did not correlate reliably with histology (data not shown). In addition, cell number comparisons between standard curves obtained using cells from 2D monolayer culture would just be approximations since the cell metabolism can change between 2D and 3D culture. Endpoint DNA analysis allows for the elimination of these diffusional and metabolic concerns. In these studies, a PicoGreen DNA assay was developed to accurately quantify cells, which allowed for the estimation of seeding efficiency and cellular proliferation of two prostate cancer cell lines within sphere-templated pHEMA scaffolds. The observed variability in seeding efficacy was expected given the nature of the capillary force cell seeding technique, but the resulting cell proliferation generated seeded scaffolds reliably containing around 1×106 cancer cells after 7-day culture. These constructs could potentially serve as the basis for biomaterial-derived tissue-engineered prostate cancer xenografts in future studies.
Conclusions
These studies demonstrate that sphere-templated pHEMA can be used as a 3D scaffold for M12 and LNCaP C4-2 prostate cancer cell culture. To seed cells within the interconnected porous structure, a capillary force-based technique was introduced that improved the cell number and distribution within the scaffolds compared to other established seeding methods. Dynamic culture conditions limited necrotic zone formation during 3D cell culture in vitro. Finally, a PicoGreen DNA assay quantitatively demonstrated the proliferation of prostate cancer cells within the 3D scaffold. The sphere-templated polymeric scaffold system and the techniques developed in these studies may have further application in the emerging field of tissue-engineered biomaterial-based cancer models.
Footnotes
Acknowledgments
This work was supported by the Nanotechnology and Physical Science in Cancer Research Training Program NIH T32CA138312 (T.J.L. and B.D.R.), TMEN-U54CA126540 (S.R.P.), PO1 CA085859 (S.R.P.), and the Veterans Affairs Research Service (S.R.P.).
Disclosure Statement
The authors confirm that there are no known conflicts of interest associated with this publication and there has been no significant financial support for this work that could have influenced its outcome.