
Abstract
Human embryo implantation involves a complex network of molecular signaling that is modulated by endocrine and paracrine pathways. Here, we performed studies using a unique and recently developed three-dimensional (3D) implantation model, characterized by an endometrium-like 3D culture system and Jar cell-derived spheroids mimicking the embryo/trophoblast. The aims were to investigate the effects of 17β estradiol (E2) and medroxyprogesterone acetate (MPA) on (1) the interaction between epithelial and stromal cells, and (2) the attachment and invasion of trophoblast cells. We observed that epithelial and stromal cells in the 3D culture were ERα+, ERβ+, and PR+. Decidualization was confirmed by enhanced prolactin gene expression on day 7 of E2 plus MPA treatment. An effect of epithelial cells on the decidualization of stromal cells was indicated by significantly higher levels of prolactin mRNA expression in the 3D culture compared to stromal cells grown within the fibrin–agarose gel matrix. On the other hand, the relative gene expressions of E-cadherin and IL-1β in epithelial cells of the 3D culture under decidualization conditions significantly differed from those in epithelial cells grown over the fibrin–agarose gel matrix without stromal cells, pointing to regulation of epithelial cells by the stroma. The attachment rate of Jar spheroids to the 3D was significantly increased by E2 plus MPA treatment. Analyses of Z-stack confocal and stained optic microscopic images demonstrated that Jar spheroids breached the epithelial cell monolayer, invaded, and were embedded into the 3D matrix in response to decidualization signals. In summary, the newly bioengineered system provides a unique model for studying interactions between the different endometrial cell compartments, via soluble-paracrine signals as well as cell-to-cell interactions, and is a useful tool to study early embryonic implantation events.
Introduction
In the human, the implantation site is not easily accessible for investigations in vivo. It appears reasonable to examine the implantation process using an in vitro model where it is possible to characterize signaling pathways through the manipulation of cells and their culture conditions, in order to recapitulate the in vivo scenario. 3 Implantation events have been investigated primarily by embryos cocultured with endometrial cells grown in a monolayer (two-dimensional, 2D), which has yielded valuable information.4–7 Nonetheless, 2D endometrial cell cultures have limitations as relevant biological properties are lost, including cellular differentiation and polarization, cell-to-cell communication, and interaction with the ECM, due to growth on artificial plastic surfaces.8–11 Several studies have demonstrated that 2D monolayers are different from 3D culture systems in structure, expression of a number of genes, and physiological responses.12–17
Therefore, a shift toward the study of implantation using three-dimensional (3D) models is occurring.18–20 The 3D model permits not only the study of cell-to-cell interactions but also the examination of cellular invasion into the ECM, and more closely resembles the in vivo scenario.8,19,21 Three-dimensional human endometrial cultures in a hydrogel matrix have been developed in a limited number of reports and some valuable information about implantation has been obtained.21–24 Therefore, the advent of 3D culture systems opened a new window for the observation of unique reproductive and developmental events of human implantation.
Although 3D models have been used for basic investigation, there are few available studies regarding long-time invasion of trophoblast cells under steroid hormone treatment. Our previous publication demonstrated that a newly developed endometrial 3D culture system supports a variety of physiological processes, such as cell polarity, cell–cell, and cell–matrix interaction, and expression of well-characterized cellular biomarkers, indicating that cells cultured in a 3D environment could better represent in vivo cellular behavior. 25 This novel 3D culture system has several significant advantages over other 3D models. First, in contrast with hydrogel culture models (i.e., using collagen and/or Matrigel as scaffold),18,19,26 the fibrin gel is not contracted by stromal cells. Second, the fibrin gel is produced using human plasma, which leads to enhanced tolerance to cells by favoring cell migration, proliferation, and secretion, and facilitates incorporation of growth factors by the fibrin gel.27–29 Third, the 3D culture constructed with established cell lines rather than with primary cultures eliminates cell heterogeneity, with high reproducibility and functional competence, and with relative low cost and easiness for preparation.25,30–32 Here, using our recently bioengineered 3D culture system model we aimed to characterize the effects of 17β estradiol (E2) and the progestin medroxyprogesterone acetate (MPA) on (1) the interaction between epithelial and neighboring stromal cells, and (2) the rates of attachment and invasion of embryo-like Jar spheroids.
Materials and Methods
Cell preparations
The human immortalized endometrial stromal cell line (HESC) and the human trophoblast Jar cell line were obtained from the American Type Culture Collection (ATCC). Ishikawa cells were provided by Dr. Lockwood, from Yale University (New Haven, CT). HESC were cultured in Dulbecco's modified Eagle's medium Ham's F-12 (DMEM/F12; Gibco) supplemented with 10% charcoal-stripped fetal bovine serum (FBS; Atlanta Biologicals), 1 mM sodium pyruvate (Gibco), and 100 U/mL–100 μg/mL of penicillin–streptomycin (Gibco). The Ishikawa cells were cultured in DMEM/F12 supplemented with 10% (v/v) FBS, 200 nM L-glutamine (Gibco), and 100 U/mL–100 μg/mL of penicillin–streptomycin (Gibco). Jar cells were cultured in RPMI 1640 (Gibco) supplemented with 10% FBS (Atlanta Biologicals), 50 μg/mL gentamycin (Gibco), 14 mM Glucose (Sigma), and 10 mM Hepes (Sigma). All cells were cultured at 37°C in a humid atmosphere with 5% CO2. Medium change occurred every other day.
Establishment of the 3D endometrial culture system
The 3D culture system was constructed as previously described by Wang et al. 25 A mixture of fibrin–agarose was used as a scaffold, with the epithelial cells (Ishikawa) on top, and the stromal cells (HESCs) residing within the matrix. Fibrin was obtained from frozen human plasma from the local Red Cross. These studies were approved by the Institutional Review Board of Eastern Virginia Medical School (IRB# 09-07-EX-0153 and IRB# 04-10-FB-0279). The effect of stromal cell concentration on the solidification of the 3D culture system was initially examined. Based on these experiments, the optimized final composition and construct set up of the 3D system was as follows: 2×106 stromal cells were suspended in 35 μL medium, and then added to 425 μL of plasma. The mixture was supplemented with 7.5 μL of 50 mg/mL tranexamic acid (Sigma) to prevent degradation of the scaffold. Type VII agarose (Sigma) was dissolved in PBS (1×) (Gibco), and 25 μL of 2% VII agarose was added into the mixture. Then, 7.5 μL of 200 mM CaCl2 (Sigma) was added to the mixture. The total volume of the mixture was 500 μL.
For measurement under microscope, 250 μL of the matrix/stromal cell construct was seeded into a well of a four-well plate (Nunc) (1.9 cm2/well) and allowed to solidify at 37°C. After 20 min, 0.5 mL of DMEM/F12 medium was added. Twenty-four hours after solidification of the 3D matrix, 7×105 Ishikawa cells in 0.5 mL of DMEM/F12 medium supplemented with FBS (10%) were seeded on top of the 3D matrix, and cultured for 3 days. Then, the established 3D culture systems were used for attachment and invasion studies.
For immunostaining and hematoxylin & eosin (H&E) staining, 500 μL of the matrix mixture was transferred to a 12-mm insert (3 μm pore size; Corning, Inc.,), and then the insert was placed inside a culture well of a 12-well cell culture plate, and allowed to solidify at 37°C. After 20 min, DMEM/F12 medium was added into the well until submerging the insert. Twenty-four hours after the 3D matrix had solidified, 4×105 endometrial epithelial (Ishikawa) cells in 250 μL DMEM/F12 supplemented with 10% FBS were seeded on the top of 3D matrix, and 1.5 mL was added into the well and co-cultured for 3 days. According to the experimental design, the 3D culture system was treated further.
Jar spheroid culture
In order to mimic an invasive trophoblast model, Jar spheroids were prepared as a published procedure 33 with minor modifications. In brief, after reaching 80% confluence, Jar cells were detached with 0.25% trypsin (Gibco). A 6×105 single-cell suspension was transferred into a 3% agarose-coated Petri dish (10 cm in diameter), and cultured in order to form small spheroids of approximately 50 μm diameter. Then, these small spheroids were transferred into an Erlenmeyer flask, and cultured for 3 days. The Erlenmeyer flask was kept on a shaker at 110 rpm. The Jar spheroids size was measured using an optic microscope equipped with a calibrated eyepiece reticle. Jar spheroids 150–200 μm in diameter were selected for studies. All cultures were in RPMI 1640 (Gibco) that was supplemented with FBS (10%) (Atlanta Biologicals), 50 μg/mL of gentamycin (Gibco), 14 mM of glucose (Sigma), and 10 mM of Hepes (Sigma) in a humid atmosphere with 5% CO2 at 37°C.
Immunohistochemistry
Immunohistochemistry was conducted to detect estrogen receptors alpha and beta (ERα, ERβ), and progesterone receptor (PR) in the cells of 3D culture system. Antibodies against ERα, ERβ, and PR (Abcam) were diluted to 1:75. Immunohistochemistry was performed as described previously. 25 In brief, the 3D co-cultures were fixed using formalin, paraffin-embedded, sectioned, and mounted on glass slides. Deparaffinization and rehydratation were followed by rinsing three times with phosphate buffer saline (PBS) for 5 min. Histochemical staining was carried out on 3D culture system sections using the DAKO kit. Negative controls were treated with a nonimmune IgG control antibody (normal mouse ascites, clone NS-1; Sigma) at similar concentration as primary antibodies. Tissue obtained by endometrial biopsy from normal volunteers under Institutional Review Board approval was used as positive controls. Representative images were photographed with an Olympus microscope using a Q-color 3 camera.
Hormone assays
In order to measure human chorionic gonadotropin (hCG), E2, and progesterone (P4) released by the Jar spheroids, Jar spheroids (150–200 μm in diameter) were inoculated into wells in a four-well plate, and cultured for 10 days. Media were collected on days 2, 4, 6, 8, and 10, and then stored at −80°C. The concentrations of hCG, E2, and P4 in the supernatant media were assayed by the Immulite hCG kit (Siemens Medical Solutions Diagnostics), and E2 and P4 kits (Siemens) on an Immulite 1000 (Siemens), respectively. For the hCG assay, the sensitivity was 1.1 mIU/mL. The inter- and intra-assay coefficients of variation were <9.9% and <5.4%, respectively. For E2, the assay sensitivity was 15 pg/mL. The intra- and inter-assay coefficients of variation were <15% and <16%, respectively. For the P4 assay, the sensitivity was 0.2 ng/mL, and the inter- and intra-assay coefficients of variation were <16% and <16%, respectively.
Analysis of the interaction between stromal and epithelial cells under decidualization conditions
Cell interactions under decidualization conditions were investigated through the expression of molecules known to be involved in implantation using real-time polymerase chain reaction (PCR). The 3D culture systems were cultured for 7 days in the presence of E2 (10−8 M; Sigma) plus MPA (10−6 M; Sigma) for decidualization. Similarly, stromal cells grown within the fibrin–agarose gel matrix (without epithelial cells) and epithelial cells grown on the fibrin–agarose gel matrix (without stromal cells) were treated with E2 (10−8 M) plus MPA (10−6 M) for 7 days and served as controls. All culture media were supplemented 5% FBS, and changed every other day.
After the 3D culture treatments, the epithelial Ishikawa cells were released from the top of the 3D constructs by 0.25% trypsin (Gibco) treatment for 4 min. Isolated Ishikawa cells on the one hand and the remaining 3D matrix with stromal cells on the other were washed in PBS, and then kept in −80°C for mRNA extraction. Similarly, Ishikawa cells growing on fibrin–agarose gel (controls) were collected using 0.25% trypsin digestion. The purity of the isolated epithelial from 3D culture system was confirmed by analysis of the specific markers cytokeratin as described in Wang et al. 25
RNA extraction, reverse transcription, and real-time quantitative PCR
Total RNA was extracted from the isolated epithelial cells and from the matrix-embedded stromal cells with the RNeasy kit (Qiagen). RNA quality was analyzed on an Agilent 2100 Bioanalyzer (Agilent Technologies) and quantification was performed on a NanoDrop spectrophotometer (Thermo Scientific).
cDNA was generated from 3 μL total RNA (≤1 μg) in 20 μL containing: 2.5 μM random hexamers, 2.5 U/μL murine leukemia virus reverse ranscriptase, 1 U/μL RNase inhibitor, 1× PCR buffer, 1 mM each deoxy-NTP, and 5 mM MgCl2 (Applied Biosystems). Reverse transcription (RT) parameters were 23°C for 10 min, 42°C for 15 min (RT reaction), 99°C for 5 min (transcriptase deactivation), and 5°C for 5 min in an iCycler thermal cycler (BioRad). cDNA solutions were then stored at −20°C. Omission of reverse transcriptase was used as negative controls, where absence of PCR products indicated a lack of contaminating genomic DNA.
Real-time RT–PCR was performed with a LightCycler Fastart DNA Master Plus SYBR green I and a LightCycler 2.0 instrument (Roche Applied Science) in a 20 μL total reaction volume, containing 2 μL cDNA as template, 4 μL master mix, 13 μL water, and 1 μL (1 μM) of each sense and antisense primers. Primers were designed using a primer designing software Primer 3. The sequence, annealing temperature, and PCR products were as follows: PRL, forward sequence 5′-AAAGCTGTAGAGATTGAGGAGCA-3′ and reverse sequence 5′-CGATTTTATGTGAATCCCTGCGT-3′, 58°C, 205 bp; E-cadherin, forward sequence 5′-CGAGAGCTACAC GTTCACGG-3′ and reverse sequence 5′-GGCCTTTTGACT GTAA TCACACC-3′, 58°C, 161 bp; IL-1β, forward sequence 5′-CACGATGCACCTGTACGATCA-3′ and reverse sequence 5′-GTTGCTCCATATCCTGTCCCT-3′, 58°C, 121 bp; and 18S, forward sequence 5′-GTAACCCGTTGAACCCC ATTC-3′ and reverse sequence 5′- GCCTCACTAAACCA TCCAATCG-3′, 58°C, 164 bp. The following LightCycler experimental run program was used: denaturation at 95°C for 10 min, and amplification program of 45 cycles (denaturation at 95°C for 10 s, annealing of primers at the specific temperature as above for 5 s and extension at 72°C for 10 s), and followed by a final extension at 72°C for 10 min. As negative controls PCR water replaced the cDNA solution (no template control). Samples were analyzed in three technical replicates and three biological replicates and normalized to 18S. Preliminary experiments allowed us to determine that expression of 18S was not affected by our treatments in the model (data not shown); therefore, 18S served as a normalization control. Relative expression levels of these genes in relation to controls were calculated using the mathematical model: ratio=2−ΔΔCP, ΔΔCP=ΔCP target (control–treatment)–ΔCP 18S (control–treatment). 34
Attachment assay of Jar spheroids to the 3D culture system
The 3D culture systems were set up into four-well plates (Nunc), and cultured with and without E2 (10−8 M) and MPA (10−6 M) for 3, 5, and 7 days. At each of those time points, 15 Jar spheroids (150–200 μm in diameter) were transferred onto the top of the 3D culture system. Jar spheroids were cocultured with the 3D culture system for attachment studies. After 25 min, the four-well plate was transferred into a bucket of a centrifuge (the top facing the bottom of the swinging bucket), and centrifuged at 1000 rpm for 5 min. The attached Jar spheroids were counted under a regular optic microscope (× 40). The attachment rate was calculated for each well as follows: the number of spheroids attached divided by the number of spheroids seeded. 25
Invasion assay for Jar spheroids into the 3D culture system
1. Labeling of Jar spheroids with CMFDA. To investigate invasion, Jar spheroids were stained with 5-chloromethylfluorescein diacetate (CMFDA, CellTracker green; Promega), a lipophilic tracer that can be introduced into live cells. Once inside the cell, this dye undergoes covalent changes restricting its passage to extracellular milieu. Labeling of the Jar spheroids was performed by incubation with the dye at a concentration of 5 μM. After 60 min of incubation at 37°C, the CMFDA dye was discarded and Jar spheroids were washed twice in PBS. Then, the Jar spheroids were incubated in culture medium at 37°C for an additional 60 min and finally washed twice with culture medium for use.
2. Invasion of Jar spheroids into the 3D culture system. The 3D culture system was set up into wells in four-well plates (Nunc) and cultured for 3 days. Thereafter, labeled Jar spheroids were transferred on to the top of 3D culture system. The 3D culture system with Jar spheroids was cultured in the presence and absence of E2 (10−8 M) and MPA (10−6 M) for 3, 5, and 7 days, and at each of those time points invasion was measured.
3. Measurement of invasion depth. The invasion depth of Jar spheroids was analyzed with Z-images captured by a Zeiss 510 Meta confocal laser scanning microscope (LSM 510; Zeiss). CMFDA was excited with a 488 nm argon laser, and followed by emission collection with a 505–530 nm band pass filter. Image acquisition was performed following optimization of LSM settings. Gain and offset in range indicator palette was optimized to enhance the visibility of image. The pinhole size corresponded to a value of 1.0 of the airy disk as calculated by the LSM software. A stack of images was collected along z-axis (Z-stacks) at an optimal thickness with 10× Plan-Neofluar objective lens NA 0.3 in a 512×512 pixel array, 8 bits/pixel. To reduce noise, each slice was scanned eight times before being averaged. Confocal z-stacks were acquired across the full invasive depth of Jar spheroid, which was the distance from the top surface of 3D culture system to invasion deepest point.
Statistical analysis
Results were obtained from a minimum of three independent experiments per experiment all performed in duplicate, and are presented as mean±SEM. p value <0.05 was considered significant. Changes in PRL mRNA expression levels were analyzed with the Friedman's Chi-Square test and the Cochran-Mantel-Haenszel statistics (based on rank scores), considering the effects of culture time and treatment, respectively. Results of gene expression changes of E-cadherin and IL-1β were analyzed with the Kruskal-Wallis test. Comparisons of attachment rates were carried out using logistic regression considering the independent effects of treatment and culture time (odds ratios). Depth of invasion of Jar spheroids was analyzed with a generalization of Friedman's Chi-Square nonparametric test.
Results
Functional characterization of the 3D culture system and Jar spheroids
We developed a human embryo implantation model for attachment/invasion studies, including an endometrium-like 3D culture system and embryo-like Jar spheroids (see schematic diagram, Fig. 1). Here, we further characterized physiological and functional properties of this implantation model. Interestingly, we observed an effect of stromal cell concentration on the solidification of the 3D matrix (Fig. 2 and Table 1). A significantly negative linear correlation was found between HESC concentration and CaCl2 concentration. This finding indicated that HESC were incorporated into the fibrin–agarose gel, and were not simply mixed into the fibrin–agarose gel. To mimic the normal endometrial tissue, 2×106 stromal cells were added into 3D matrix in all experiments.
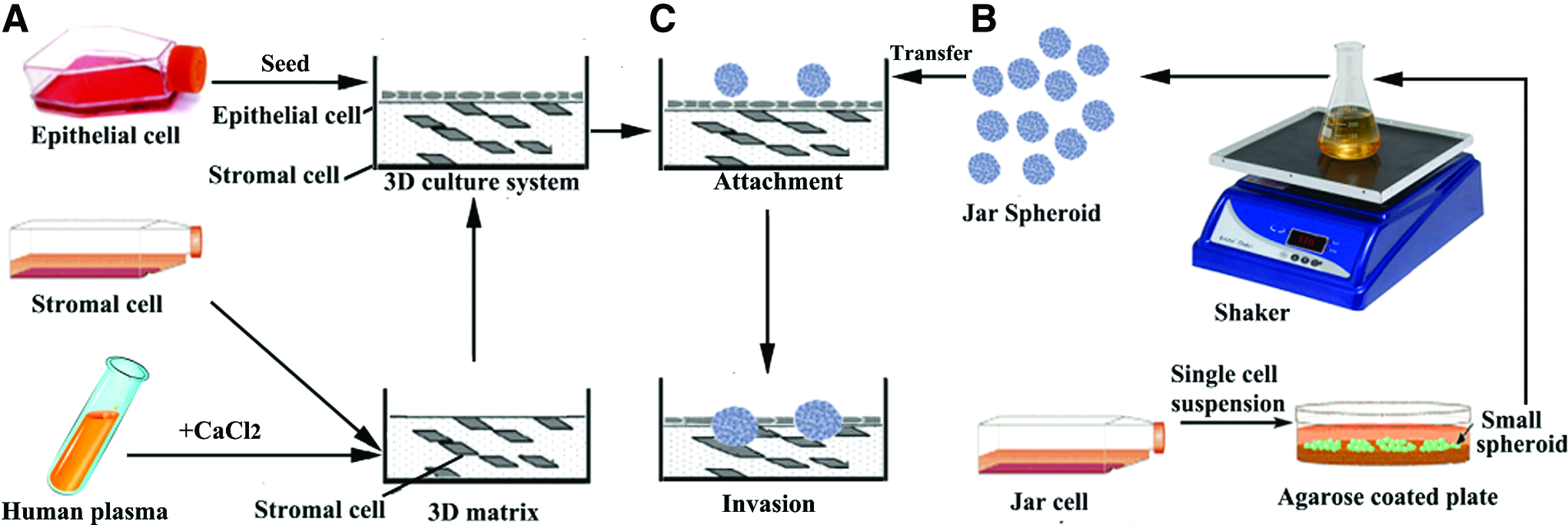
Schematic diagram of the development of the three-dimensional (3D) endometrial culture system.

Effect of human immortalized endometrial stromal cell (HESC) concentration on the solidification of the 3D matrix. Bars represent the final concentration of CaCl2 needed for solidification of the 3D matrix under different HESC concentrations. The 3D matrix mixture was solidified within 20 min at 37°C. The relationship coefficient, R2, showed that there is a significantly negative linear correlation between HESC concentration and CaCl2 concentration.
We studied the effect of HESC concentration on the solidification of the 3D matrix. Without stromal cells in the (500 μL) 3D matrix, the matrix solidification required more than 11.7 mM CaCl2. With an increase in the HESC concentration, CaCl2 concentration showed a tendency to decline. When 500 μL 3D matrix contained 2×106 HESCs, the 3D matrix solidification only required 3.0 mM CaCl2. HESC, human immortalized endometrial stromal cell; 3
The expression of ER and PR in the cells of the 3D culture system was evaluated by immunohistochemistry (Fig. 3). Ishikawa cells and HESCs within the 3D culture system expressed ERα+, ERβ+, and PR+, implying that the 3D culture system can respond to estrogen and progestin stimulation. Negative controls included sections that were treated with a nonimmune IgG control antibody.

Immunohistochemical analyses of progesterone receptor (PR) and estrogen receptor (ER) in cells present in the 3D culture system (Ishikawa endometrial epithelial cells and endometrial stromal cells (HESC).
To assess functional properties of the trophoblast Jar spheroids, we investigated hormone secretion in the supernatant medium. Figure 4A and B shows that the secretion of hCG, E2, and P4 by Jar spheroids increased with culture time. The secretion of hCG, E2, and P4 into the culture medium was measured as per spheroid at 48-h interval. On day 10 of culture, one Jar spheroid secreted 30.7±6.30 mIU of hCG, 4.5±0.32 pg of E2, and 2.1±0.30 ng of P4, during a 48-h period of culture. The results revealed that Jar spheroids can produce hormones that are associated with implantation. Figure 4C presents a representative light microphotograph of Jar spheroids during culture.
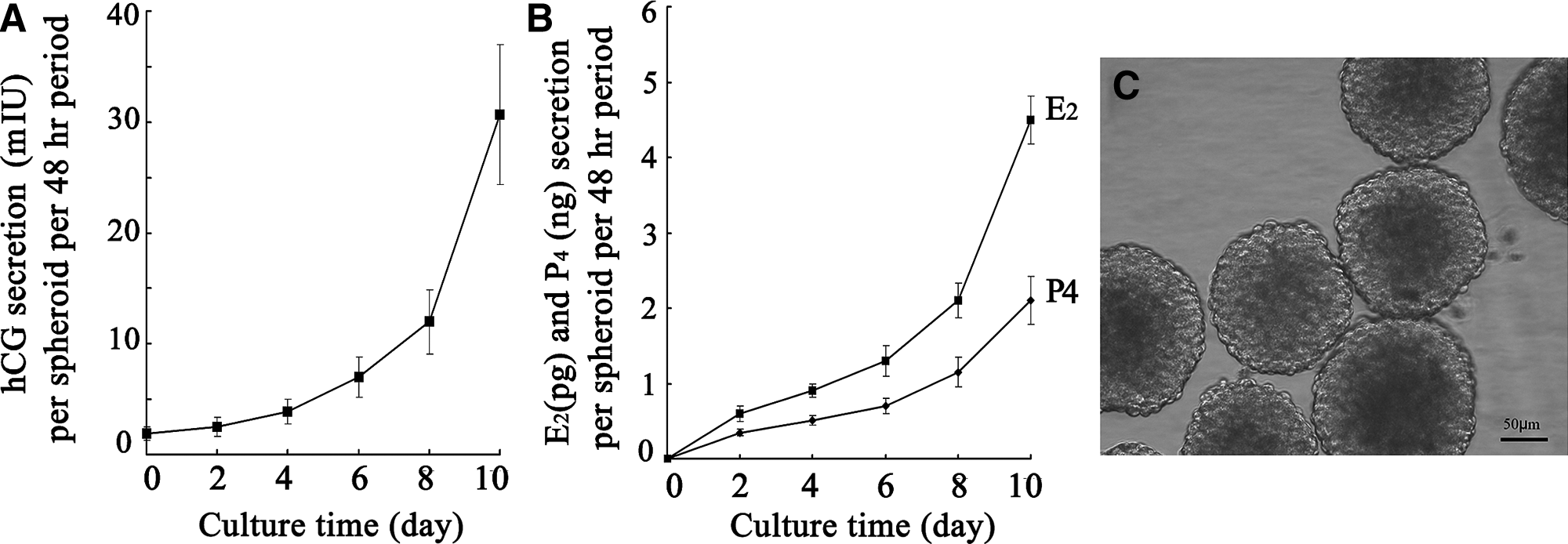
Hormone secretion by Jar spheroids and their morphological appearance.
Interaction between epithelial cells and stromal cells in the 3D culture system
We investigated the effect of epithelial cells on the decidualization of HESCs in the 3D culture system by analyzing the gene expression of PRL, a decidualization marker. We compared results of HESC grown within the fibrin–agarose gel alone (and without epithelial cells) with those of HESCs from the 3D culture system (complete system with stromal and epithelial cells) in the presence and absence of E2 (10−8 M) plus MPA (10−6 M) on days 3, 5, and 7. The results of PRL mRNA relative expression are shown in Figure 5. Overall, there was a significant treatment effect (p=0.03). PRL mRNA expression did not show a temporal change in HESCs grown within the fibrin–agarose gel and in the 3D cultures without steroid treatment. However, the relative expression of PRL mRNA in HESC significantly increased under E2 plus MPA treatment on day 7, confirming decidualization transformation. The relative gene expression level of PRL in decidualized HESCs in the 3D culture was significantly higher than in decidualized HESCs grown within the fibrin–agarose gel (p<0.05), indicating that epithelial cells were involved in the decidualization of HESCs in the simulated endometrium, and suggesting a facilitating or synergistic paracrine interaction.
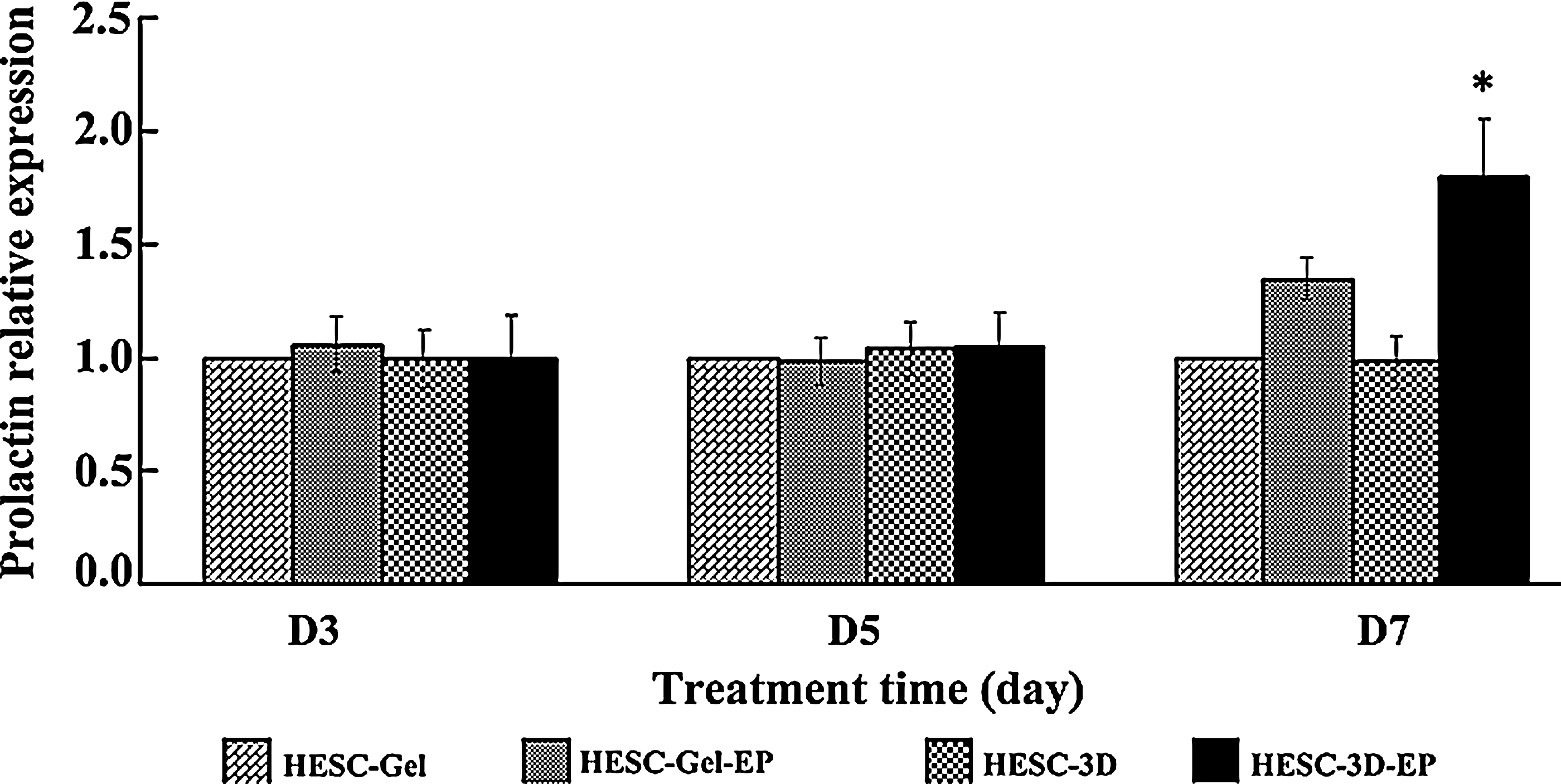
Decidualization of HESC under E2 plus medroxyprogesterone acetate (MPA) treatment. The relative mRNA expression of PRL in the HESC of the 3D culture system (Ishikawa cells and stromal cells/matrix) compared to HESC grown within the fibrin–agarose gel (without Ishikawa cells) in the presence and absence of E2 (10−8 M) plus MPA (10−6 M) on day 3, 5, and 7. Results are presented following 18S normalization and setting their individual level of expression in HESC grown within the fibrin–agarose gel without E2 plus MPA treatment arbitrarily as 1. Data are presented as mean±SEM, n=3 independent experiments. A significant overall treatment effect was observed (p=0.03). *p<0.05 comparing HESC-3D-EP versus other conditions on day 7. HESC-Gel: HESC grown within the fibrin–agarose gel without E2 plus MPA treatment; HESC-Gel-EP: HESC grown within the fibrin–agarose gel with E2 plus MPA treatment; HESC-3D: HESC from 3D culture system without E2 plus MPA treatment; HESC-3D-EP: HESC from 3D culture system with E2 plus MPA treatment.
An effect of HESCs on the epithelial (Ishikawa) cells was also observed in the decidualized 3D culture following 7 days of treatment. As can be seen in Figure 6A for E-cadherin there was an overall effect of treatment on gene expression (p=0.03). Its relative expression level was significantly decreased in the Ishikawa cells from the decidualized 3D culture system compared to Ishikawa cells cultured on the fibrin–agarose 3D gel (constructed with matrix alone and epithelial cells but without stromal cells) with/without E2 plus MPA treatment (p<0.05). Similarly, there was an overall effect of treatment on gene expression (p=0.02) for IL-1β. Its relative mRNA expression was downregulated in Ishikawa cells from the decidualized 3D culture system compared to Ishikawa cells cultured on the fibrin–agarose 3D gel (matrix alone) with (p<0.05) and without (p<0.04) E2 plus MPA treatment (Fig. 6B). Furthermore, the expression of these genes in Ishikawa cells from the 3D culture system without E2 plus MPA treatment were downregulated compared to Ishikawa cells cultured on fibrin–agarose gel without E2 plus MPA treatment. These effects are consistent with the concept that the expression of certain genes in the epithelial cells of the 3D culture system was associated with HESC paracrine signaling, in addition to steroid hormone regulation.
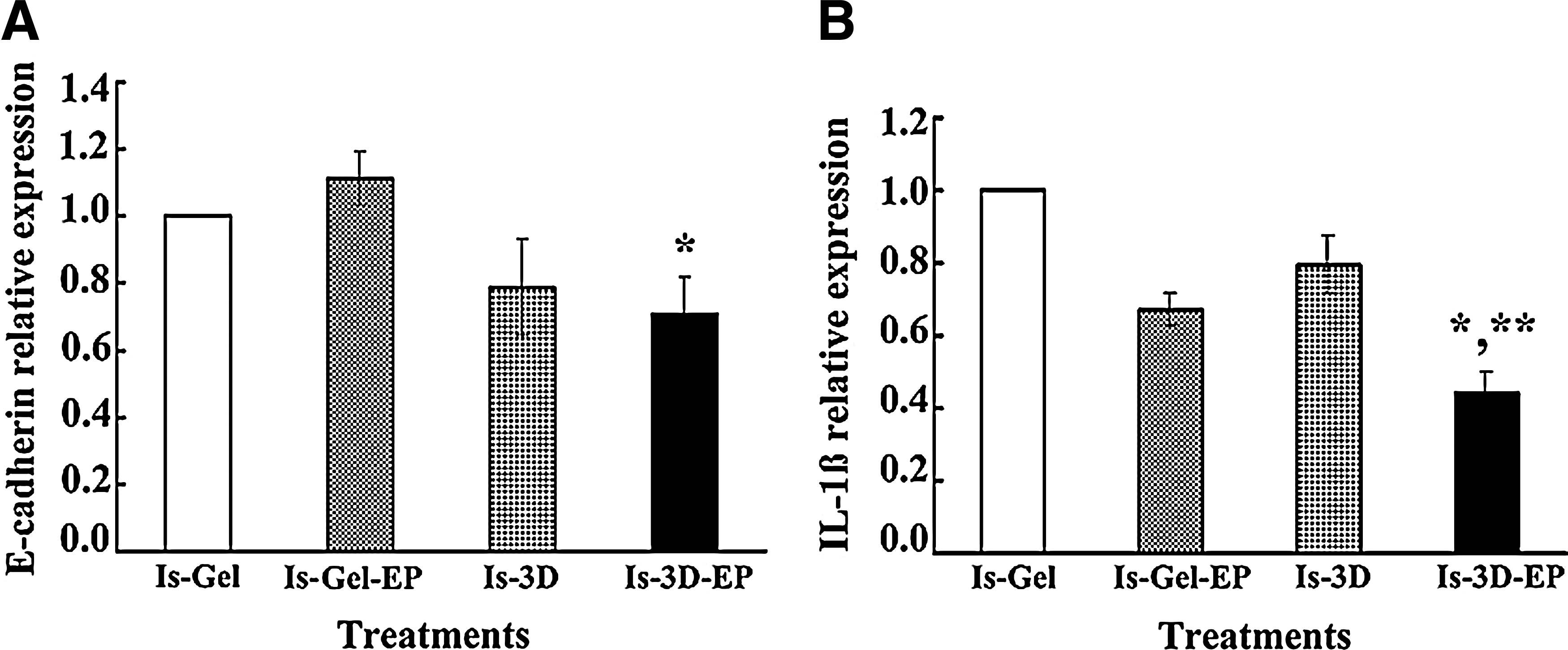
The relative mRNA expression of E-cadherin and IL-1β in epithelial cells (Ishikawa cells) of the 3D culture system and in Ishikawa cells cultured on a fibrin–agarose gel (matrix alone) in the presence and absence of E2 plus MPA. The 3D culture system and Ishikawa cells on a fibrin–agarose gel were cultured in the presence and absence of E2 (10−8 M) plus MPA (10−6 M) for 7 days. The relative expression levels of E-cadherin and IL-1β in Ishikawa cells were analyzed on day 7. Results are presented following 18S normalization and setting their individual level of expression in Ishikawa cells on the fibrin–agarose gel without E2 plus MPA treatment arbitrarily as 1. Data are presented as mean±SEM, n=4 independent experiments.
Effect of E2 plus MPA treatment on receptivity of the 3D culture system
The effect of steroid treatment on the receptivity of the 3D culture system was analyzed by the attachment rates of Jar spheroids to the 3D culture in the presence and absence of E2 (10−8 M) plus MPA (10−6 M). There were significant overall treatment and time effects as shown in Figure 7A. The p value for the treatment effect was 0.0003, indicating higher the odds of attachment in the treatment group. In addition, the p value for time effect was 0.003, which indicated that the odds of attachment differed between days. Furthermore, there was a significant difference between days 3 and 7 of culture (p=0.009). These results showed that E2 plus MPA treatment increased the receptivity of 3D culture system. Figure 7B shows a Jar spheroid attached to the Ishikawa monolayer on top of the 3D construct.
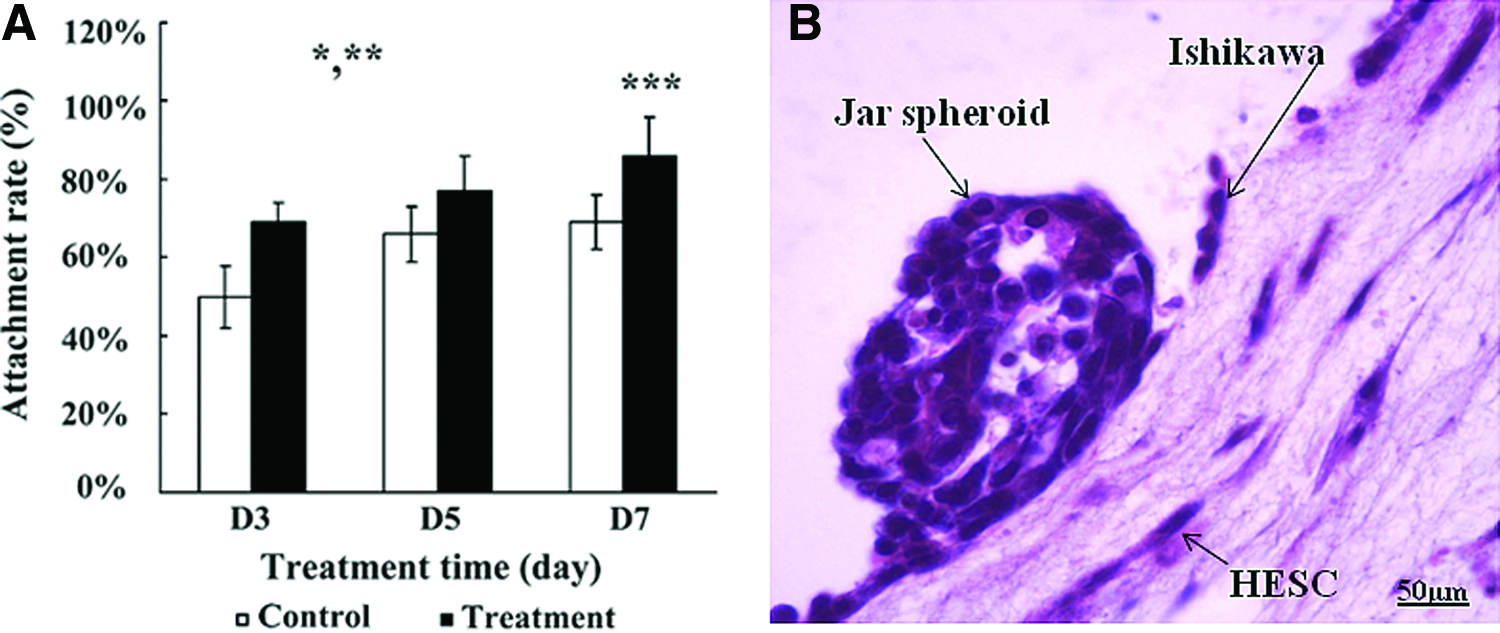
Attachment of Jar spheroids to the 3D culture system in the presence and absence of E2 and MPA treatment.
Effect of E2 plus MPA on the invasion of Jar spheroids into 3D culture system
We measured the invasion depth of Jar spheroids into the 3D matrix in the presence and absence of E2 plus MPA using confocal microscope by obtaining z-images. As depicted in the schematic illustration (Fig. 8A) and in real-time confocal images (Fig. 8B), the Jar spheroids were labeled by CMFDA and transferred onto the top of the 3D culture system, and the labeled Jar spheroid-3D cocultured system was cultured in the presence and absence of E2 (10−8 M) plus MPA (10−6 M) for 3, 5, and 7 days. There were significant overall effects of treatment (p=0.0007) and time (p=0.007) regarding invasion depth, with depth of invasion increasing from days 3 through 7, and higher under treatment conditions than in controls (Fig. 8C). In addition, we stained slides (H&E) from 3D cultures fixed with formalin to evaluate the invasion of Jar spheroids under E2 plus MPA treatment. Slides from 10-day cocultured Jar spheroids and the 3D culture system under E2 plus MPA treatment unequivocally showed that the Jar spheroid breached the epithelial cell monolayer on the top of 3D culture system and digested the 3D matrix, and then buried into 3D culture system completely (Fig. 8D, E). However, the case of complete invasion was not observed without E2 plus MPA treatment. These results indicated that the decidualiztion transformation in 3D culture and enzyme secretion by Jar spheroid play major roles in the process of invasion.
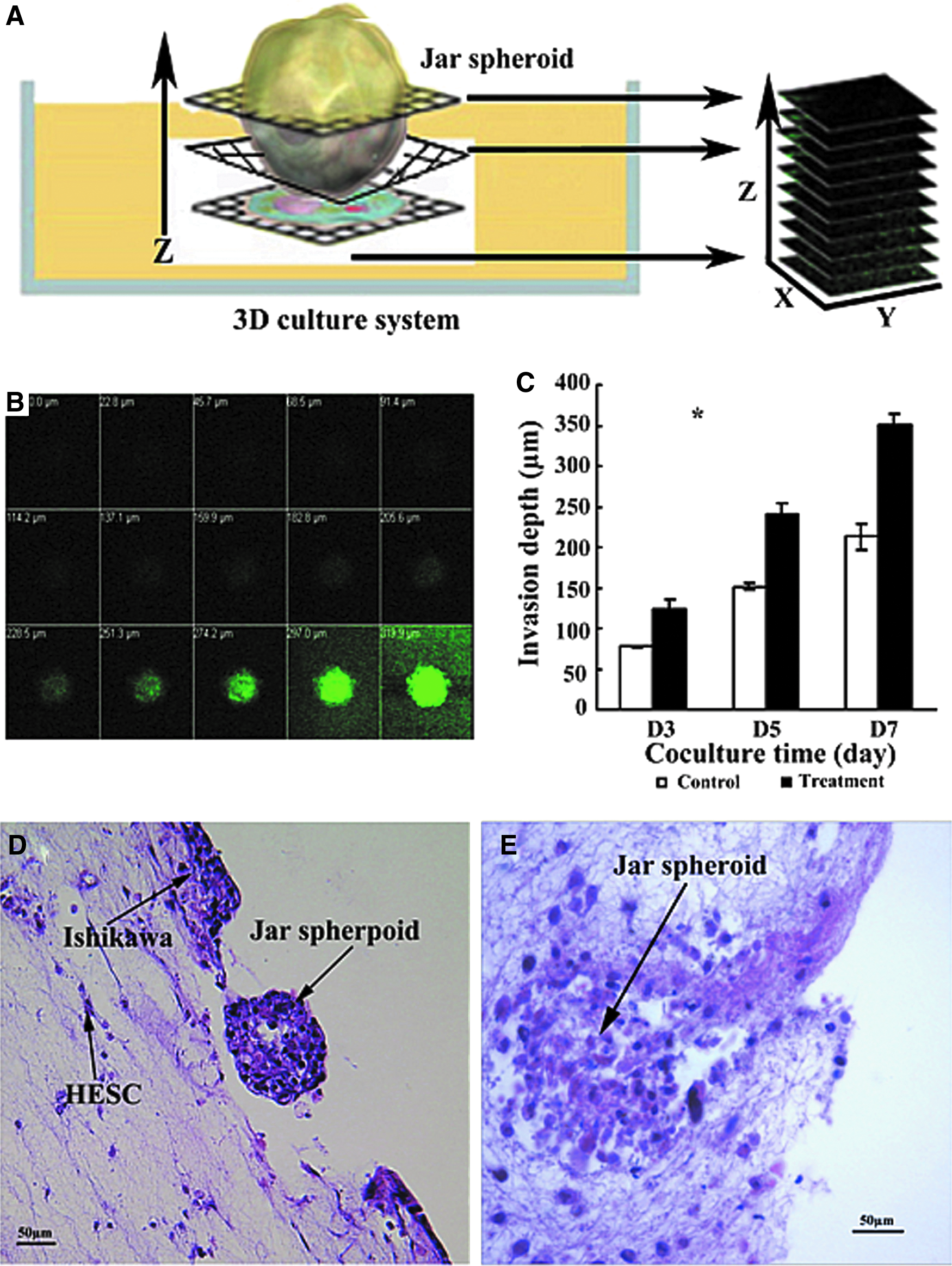
Invasion of Jar spheroids into the 3D culture system in the presence and absence of E2 plus MPA treatment.
Discussion
In our previous report 25 we characterized the architecture, growth, and survival of the 3D endometrial culture system, as well as the attachment of embryo-like Jar spheroids. Moreover, the epithelial/stromal cells phenotypes and the immunolocation of αvβ3 integrin in the 3D culture were confirmed. Here, we further characterized physiological and functional properties of the 3D model. We confirmed that the epithelial and stromal cells residing in the 3D culture system express ERα, ERβ, and PR, as previously reported by Meng et al., 35 and that Jar spheroids secrete hCG, E2, and P4 as reported earlier. 36 Furthermore, we demonstrated reciprocal interactions between epithelial and stromal cells residing in the 3D model in a possible paracrine manner, and increased rates of attachment and invasion of embryo-like Jar spheroids in response to steroid decidualization signals.
We observed that the need for CaCl2 concentration decreased with an increase of stromal cell concentration in 3D matrix solidification, which appears to differ from other 3D models, such as those using a collagen matrix.18,19 This indicated that the stromal cells really took part in the process of 3D matrix solidification as an organic component, and were not simply mixed into the fibrin–agarose gel. In such endometrium-like 3D culture system, it is conceivable that the transformation of stromal cells to decidual cells certainly results in the structurally and functionally relevant transformation of 3D culture system to create an appropriate microenvironment for implantation. Moreover, the expression patterns of ERα, ERβ, and PR in the epithelial and stromal cells shown herein were of the same kind as those found in endometrial tissues in vivo. These attributes provide optimization for the establishment of long-term and physiological 3D culture systems.
Multicellular Jar spheroids hold a particular advantage over monolayer cultures of trophoblast cells for the study of implantation, particularly when associated with a 3D endometrial structure. In our present studies, we demonstrated that the Jar spheroids have the ability to produce E2, P4, and hCG, 36 indicating that Jar cells within spheroids are physiologically active. It is well established that E2, P4, and hCG play autocrine/paracrine roles in trophoblast invasion during implantation. 37 hCG has direct effects on the endometrium and mediates cross talk between the embryo and the epithelium through hCG receptors. 38 Therefore, trophoblast Jar spheroids represent an appropriate embryo-like model for studies mimicking implantation.
We evaluated signs of functional decidualization of the endometrium-like 3D culture system by analyzing the expression of the decidual marker PRL. We observed that the decidualization of the 3D culture was greatly facilitated by cocultured epithelial cells, with evidence that the PRL gene expression of stromal cells in the 3D was higher than that of stromal cells grown within the fibrin–agarose gel matrix (without Ishikawa cells) under similar conditions. Similarly, in the rat, the removal of uterine epithelium interferes with stromal decidualization. 39 Therefore, this model may mimic the normal endometrial environment more accurately than monolayer cell cultures. In addition, these data suggested that stromal decidualization in the human is under dual regulation. The first pathway is mediated by a direct effect of serum hormones, such as sex steroids, on stromal cells, and the other is an effect via paracrine signaling.
In addition to the effect of epithelial cells on the decidualization of stromal cells, we documented an effect of stromal cells on the transcript expression of adhesion and inflammatory molecules in epithelial cells of the 3D under decidualization conditions. It is well known that the roles of cycle-dependent adhesion molecules and inflammatory factors are strongly relevant to implantation.40,41 The expression of these various molecules undergo spatial and temporal changes within the cycling endometrium.42–44
E-cadherin is a class of cell adhesion molecules, and generally thought to inhibit the attachment/invasion of embryo. However, its role in endometrium is still unclear during the receptive phase. Here, we analyzed the gene expression of the adhesion molecule E-cadherin in the 3D culture system. E-cadherin in the epithelial cells of 3D culture system was significantly downregulated under decidualization signaling, whereas it was upregulated in epithelial cells grown on the fibrin–agarose gel (without HESC) in response to estrogen plus progestin stimulation. To the best of our knowledge, this is the first indication that decidualization signals from endometrial stromal cells may downregulate the expression of E-cadherin in endometrial epithelial cells. However, the results from different studies on E-cadherin expression in endometrium are not consistent in the literature.45–47 Our finding supported the hypothesis that E-cadherin is downregulated to facilitate the dissociation of epithelial cells during blastocyst invasion in the secretory phase. 47
IL-1β is a multi-functional cytokine in the endometrium 48 critical to the generation of an inflammatory response by the endometrium, and also plays a role in receptivity and implantation. 44 We observed that the gene expression of IL-1β in epithelial cells of the 3D model was significantly downregulated in response to estrogen plus progestin stimulation, indicating that stromal cells can downregulate IL-1β expression of cocultured epithelial cells through paracrine pathways. Cocultured stromal cells regulating the expression of IL-1β in epithelial cells may reflect an endogenous regulatory action by which the inflammatory response of epithelial cells is decreased to facilitate the receptive state of the endometrium.
Based on the analyses of decidualization, adhesion molecules, and inflammatory factors, we concluded that the 3D model mimics more closely the in vivo microenvironment than monolayer endometrial cells, and therefore provides a functional and structural model to investigate the activities of genes relating to implantation. In addition to the structure and the interaction of epithelial–stromal cells within the 3D model, more relevant functional characteristics were observed in the 3D model coupled with Jar spheroids, as evidenced by attachment and invasion of Jar spheroids into the matrix.
The adhesion of the trophoblast to the epithelium is one of the initial key events of human embryo implantation.49,50 In the current Jar spheroid-3D cocultured system adhesion assay, we observed that the receptivity of 3D culture system to Jar spheroids was enhanced significantly by estrogen and progestin treatment, confirming the concept that these sex steroids are critical for the development of an appropriate environment for blastocyst implantation.
The process of trophoblast invasion is characterized by a controlled trophoblast cell penetration of the luminal epithelial layer, invasion of the endometrial stroma, remodeling of the maternal vasculature, and development of the placenta.51–52 For successful invasion, trophoblast cells induce expression of a number of genes involved in digestion of the ECM.53,54 In the present study, analysis of Z-images for trophoblast invasion revealed increased depth of invasion on a time- and E2 plus MPA treatment dependency. Moreover, immunohistochemcial images unequivocally demonstrated that trophoblast cells breached the epithelial cell monolayer, and migrated into the matrix, and that estrogen plus progestin significantly facilitated the process of invasion with degradation of the 3D matrix under the areas of spheroids attachment. These observations may support the hypothesis that trophoblast cells secrete factors such as metalloproteases, enzymes known to degrade ECM of the uterine tissue into which the trophoblast invades. On the other hand, the endometrium limits trophoblast invasion by regulation of degradation via the production of specific inhibitors, such as tissue inhibitor of metalloproteases,52–55 secreted both by trophoblast cells and decidual stromal cells.18,56 Taken together, our findings demonstrated that sex steroids regulated the invasion of trophoblast cells through these pathways of decidualization transformation in endometrium, enzyme secretion by trophoblast cells, and the growth of trophoblast. Therefore, we believe that this model has significant potential for the characterization of key events associated with embryo invasion.
In summary, the development of a reproducible and functionally competent endometrial 3D culture system combined with multicellular Jar spheroids enabled us to monitor the processes of attachment and invasion in real-time and high resolution using reverse and confocal microscope. Importantly, the model will allow quantitative measurement of attachment efficiency, the invasion depth, and the interaction with epithelial and stromal cell compartments. We demonstrated that estrogen and progestins are stimulants of embryo attachment/invasion to the simulated endometrium. They appeared to influence gene expression by a direct regulation of epithelial and stromal cell acting through their respective receptors, and also by an indirect effect via paracrine pathways. We conclude that this 3D model (1) will allow for identification of molecules specifically responsible for paracrine mediation, as well as further characterization of cell-to-cell interactions, and (2) is a useful tool to study the orderly sequence of events leading to implantation, with applications for both profertility and contraceptive clinical research.
Authors' Contribution
H.W. performed all the experiments presented herein; S.A. collaborated in immunofluorescence studies; L.Y. collaborated in real-time PCR studies; B.R. performed the statistical analysis; J.H. contributed to manuscript preparation; S.B. and S.O. contributed to the study design, data interpretation, and manuscript preparation/review.
Footnotes
Acknowledgments
We are thankful to Dr. Frank Lattanzio from the Department of Physiological Sciences and Ophthalmology, and Dr. Earl Godfried from the Department of Pathology and Anatomy at Eastern Virginia Medical School for assistance in confocal microscopy. This project was funded by a grant and a Serono Endowment from the Howard and Georgeanna Jones Foundation for Reproductive Medicine to S.O. and S.B. The funding sources had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Disclosure Statement
No competing financial interests exist.