
Abstract
Adult newts (Notophthalmus viridescens) are capable of complete lens regeneration that is mediated through dorsal iris pigment epithelial (IPE) cells transdifferentiation. In contrast, higher vertebrates such as mice demonstrate only limited lens regeneration in the presence of an intact lens capsule with remaining lens epithelial cells. To compare the intrinsic lens regeneration potential of newt IPE versus mouse lens epithelial cells (MLE), we have established a novel culture method that uses cell aggregation before culture in growth factor-reduced Matrigel™. Dorsal newt IPE aggregates demonstrated complete lens formation within 1 to 2 weeks of Matrigel culture without basic fibroblast growth factor (bFGF) supplementation, including the establishment of a peripheral cuboidal epithelial cell layer, and the appearance of central lens fibers that were positive for αA-crystallin. In contrast, the lens-forming potential of MLE cell aggregates cultured in Matrigel was incomplete and resulted in the formation of defined-size lentoids with partial optical transparency. While the peripheral cell layers of MLE aggregates were nucleated, cells in the center of aggregates demonstrated a nonapoptotic nuclear loss over a time period of 3 weeks that was representative of lens fiber formation. Matrigel culture supplementation with bFGF resulted in higher transparent bigger-size MLE aggregates that demonstrated increased appearance of βB1-crystallin expression. Our study demonstrates that bFGF is not required for induction of newt IPE aggregate-dependent lens formation in Matrigel, while the addition of bFGF seems to be beneficial for the formation of MLE aggregate-derived lens-like structures. In conclusion, the three-dimensional aggregate culture of IPE and MLE in Matrigel allows to a higher extent than older models the indepth study of the intrinsic lens-forming potential and the corresponding identification of lentogenic factors.
Introduction
R
Methods
Generation of primary newt IPE cells and aggregate culture
To generate primary newt IPE cell cultures, seven newts were anesthetized by 0.5% Ethyl 3-aminobenzoate methane sulfonate in 1× phosphate buffer saline (Sigma-Aldrich, St. Louis, MO) and sacrificed for surgical removal of eyeballs. Eyeballs were placed on calcium and magnesium-free Hanks' balanced salt solution, followed by removal of the dissection of dorsal and ventral iris cells as previously published. 7 Newt IPE cells were cultured for 2 weeks on collagen-coated plates in regular culture medium (L15 medium, 65% dilution from original, containing 100 μg/mL kanamycin sulfate and 2.5 μg/mL Amphotericin B solution, and 10% Fetal bovine Serum (FBS)) and treated by 3.75 U/mL dispase overnight at room temperature. After cell detachment, cells were transferred to 1.5 mL Eppendorff tubes containing 2000–3000 cells per tube. Cells were pelleted by centrifugation at 100 g (proportional 1000 rpm) for 2 min, washed several times to remove residual dispase, and incubated at 27°C for 2 days in L-15 culture medium for IPE cell aggregation.
For Matrigel culture, tissue culture dishes (35 mm) or six-well plates were coated with 100 μL of Growth factor-reduced Matrigel (cat#354230; BD Biosciences, San Jose, CA) in form of a gel drop and kept for 2 h at 27°C for gel formation. IPE aggregates were transferred inside of the gel drop followed by the addition of 2 mL L-15 cell culture medium. Medium was changed every 3 days.
For suspension culture, IPE aggregates were transferred into tissue culture-treated 24-well plates followed by the addition of L15 culture medium (control) or L15 culture medium supplemented with 100 ng/mL bFGF (Peprotech Rocky Hill, NJ). Media were changed every 3 days.
Generation of primary MLE and mouse IPE cells and aggregate culture
After mouse lens excision, lenses were submerged in Advanced Minimal Essential Medium supplemented with 20% FBS and 1 × antibiotics/antimycotics (MEM/20%FBS, regular culture medium; Gibco®, Life Technologies, Grand Island, NY), and the posterior capsule was opened by making three clockwise incisions. The capsule was peeled from the lens fiber cell mass, and the anterior capsule was pinned with the MLE cells facing the bottom of a 3 mm culture dish using six entomological pins (D1, Watkins and Doncaster, Kent, United Kingdom). Residual lens fibers were removed by changing the medium. After 1-week culture in regular culture medium, lens capsular bags were removed, and MLE were further expanded. Primary mouse IPE cells were derived from eYFP mice bred from the ES cell line YC5 (stock #011982-MU) obtained from the Miami University Breeding Colony of Dr. Michael L. Robinson, PhD. In brief, after iris dissection, cells were dissociated by trypsinization and culture expansion in MEM/20%FBS. Cells were used at passage 5 for aggregation according to the protocol for MLE.
For generation of MLE aggregates, cells of passage 10 to 14 were trypsinized followed by the determination of cell counts and addition of, for example, 5000, 35,000, 50,000, 100,000, and 500,000 cells to 1.5 mL Eppendorff tubes by adjusting the fill volume to 1.5 mL with regular culture medium. For aggregation MLE cells were pelleted at 2000xg for 5 min followed by overnight incubation at 37°C in a CO2 incubator. For gel formation, each well of a 24-well cell culture plate was coated with 300 μl growth factor-reduced Matrigel or human collagen type I (cat#354265, BD Biosciences, San Jose, CA) followed by 20 min incubation at 37°C in a CO2 incubator by keeping the plate in a tilted state. Two to three aggregates were pipetted with a p1000 pipette into the middle of the Matrigel, and incubated for 30 min without medium followed by the addition of regular culture medium (control) or regular culture medium supplemented with 100 ng/mL bFGF (murine FGF-basic; cat#450-33 PeproTech, Inc., Rocky Hill, NJ). Aggregates were observed at different time points ranging between 0 and 3 weeks of culture.
Histology and immunohistochemistry
IPE aggregates were removed from the gel and fixed with 4% paraformaldehyde in PBS overnight 4°C. For histology analysis, aggregates were dehydrated using an ethanol-xylene gradient followed by paraffin embedding for sectioning. Tissue sections of 15 μm thickness were transferred onto gelatin-coated slides for analysis by immunohistochemistry. After tissue rehydration, samples were washed with PBS and PBST (PBS with 0.2% Triton X-100), followed by blocking with 10% Goat serum in PBS, followed by αA-crystallin antibody (supernatant of Hybridoma culture medium, nondiluted) 8 overnight at 4°C. For the secondary antibody, 1:100 anti-mouse IgG conjugated with Alexa488 (Life Technologies) was used, at 2 h RT. Selective sections were rehydrated and stained with Hematoxylin followed by dehydration (50% to 95% ethanol) and staining with Eosin.
MLE aggregates were fixed for 30 min in 2% paraformaldehyde 0.1% Triton X-100. For histological analysis, aggregates were dehydrated using an ethanol-xylene gradient. Due to the small size and invisibility of aggregates, aggregates were pipetted along with small amounts of xylene into histology boats using a p1000 pipette. After evaporation of the xylene, paraffin was added so that the aggregate was positioned in the very front of the paraffin specimen. Aggregate sections of 15 μm were mounted onto X-tra® (Leica Biosystems, Buffalo Grove, IL) microscope slides, followed by removal of paraffin and staining with 4 μg/mL Hoechst stain (33258, [Molecular Probes®; Life Technologies]) for nuclear detection. After fluorescence analysis, selective sections were chosen for additional Hematoxylin/eosin staining. Imaging was performed using a BX51 microscope (Olympus, Center Valley, PA) under fluorescence setting with a CCD camera (RTKE Spot; Diagnostics Instruments, Inc., Sterling Heights, MI) and imaging software (Spot software version 4.1, Diagnostics Instruments, Inc).
Immunohistochemistry included staining of the fixed complete aggregates with primary antibody and βB1-crystallin (β-crys (FL-252), sc-22745) at 1:250 dilution overnight followed by staining with secondary antibody coupled to cy3 (Jackson Immunoresearch) at 1:600 dilution and staining with TOTO-3 dye (T3640, Molecular Probes; Life Technologies) for 2 h. Samples were analyzed with an Olympus Fluoview™ 1000 confocal microscope under sequential settings. A Kalman filter was used for elimination of background interference. For mid-section imaging, samples were analyzed using a 20× objective under phase-contrast transmission and cy3 settings by taking an average of three scans in 1 μm intervals over 10 μm total within the middle of the aggregate.
MLE aggregate densitometry and area measurements
Phase-contrast images of MLE aggregates were analyzed by NIH ImageJ 1.43s by measuring the mean intensity (MI) from same-size area rectangles spanning from the outer aggregate edge toward the center representative for increases in transparency. At least seven samples/groups were used for analysis. Statistical analysis: Two-way ANOVA. p<0.05 was used as a criterion for significance.
Average aggregate areas [mm2] were determined by tracing the aggregate outline from corresponding phase-contrast images using NIH ImageJ 1.43s. The measured area pixels were converted into mm by use of a standard size. At least seven samples/groups were used for analysis. Statistical analysis: Two-way ANOVA. p<0.05 was used as a criterion for significance.
Protein extraction
Proteins were extracted with the NE-PER® Nuclear and Cytoplasmic Extraction Kit under modified conditions (Pierce Biotechnology, Rockford, IL) by using a combined lysis buffer containing 25 μL Cytoplasmic Extraction Reagent 1 (CERI), 1 μL CERII, and 12.5 μL Nuclear Extraction Reagent (NER)/sample. To obtain sufficient protein extract from MLE aggregates for immunoblotting, each sample consisted of protein extracts from 6 to 12 aggregates that were pooled together. Protein extracts were frozen at −20°C until further use.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis and western-blotting
Due to the small sample size, the corresponding aggregate protein extracts were loaded completely for sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (10% SDS gels). Proportional 20 μg total protein of whole lens and lens fiber, as well as 500 ng Bovine β-H crystallin (cat#SPP-235; Stressgen, Victoria, BC) in combined NE-PER lysis buffer were loaded as controls for β-crystallin expression. After Western transfer to PVDF membranes, membranes were incubated with the primary polyclonal antibodies for βB1-crystallin (β-crys (FL-252): sc-22745), aquaporin 0 (AQP0 (H-44):sc99059) α-smooth muscle actin 1 (αSM1: sc-130617), caspase-3 (caspase-3 p11 (C6): sc-271759), and β-tubulin (β-tubulin (H-235): sc-9104) as a loading control (Santa Cruz Biotechnology, Inc., Santa Cruz, CA). Staining with primary antibodies was followed by alkaline phosphatase-coupled secondary antibodies (Santa Cruz Biotechnology, Inc.) at 1:5000 dilutions and staining with enhanced chemifluorescence Western Blotting Kit (Amersham, Piscataway, NY). Membranes were scanned with a Biospectrum 500 Imaging System with an LM26 and Biochemi500 Camera f/1.2 and Vision Works LS software. Antibody staining intensities were measured using NIH ImageJ 1.43s. Densitometric analysis included measurement of same-size areas from experimental and β-tubulin (loading control) detected bands divided by the corresponding same-size area background measurement taken below the bands of interest. The corresponding experimental value was divided by the value obtained for the β-tubulin loading control for normalization. To normalize changes in expression, the average of three 0 time point measurements was set to one-fold. Three samples per time point (t0, 1wk, and 2wk) were analyzed. Statistical analysis: One-way ANOVA with Bonferroni post-test and Two-way ANOVA, p<0.05 was used as a criterion for statistic significance.
All animal studies were approved by the University of Dayton Laboratory Animal Institutional Review Board.
Results
Matrigel promotes complete lens formation of newt IPE aggregates
To study the necessary requirements for induction of newt IPE-dependent lens formation in different culture systems, proportional 2000–3000 dorsal or ventral IPE cells were aggregated followed by transfer to regular tissue culture dishes (suspension culture) or growth factor reduced Matrigel (Matrigel culture) and the addition of regular L15 culture medium.
While ventral IPE aggregates grown in suspension culture did not demonstrate any lens formation within 2 weeks and remained similar in size with a rounded pigmented appearance (Supplementary Fig. S1Ac, d; Supplementary Data are available online at
bFGF, basic fibroblast growth factor.
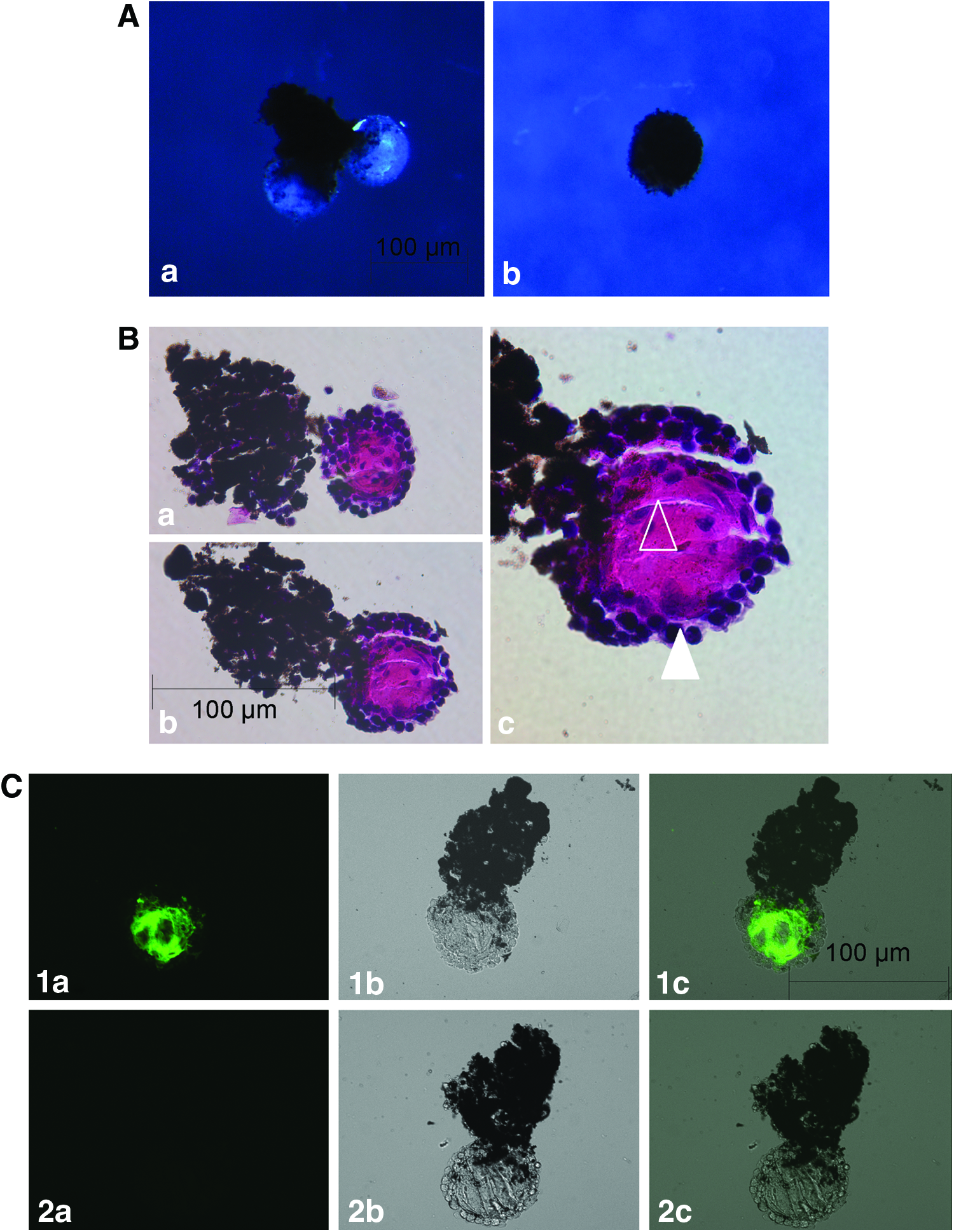
When cultured in Matrigel® the bulk part of dorsal and ventral aggregates remained pigmented and did not show any changes in aggregate shape or size. After 7 to 14 days in culture, dorsal IPE aggregates demonstrated appearance of outward growing lens structures in 22 out of 24 aggregate; while no lens formation was observed in ventral IPE aggregates (Fig. 1 and Table 1). At day 17, dorsal aggregates demonstrated complete lens formation (Fig. 2Aa) versus no signs of lens formation in ventral aggregates (Fig. 2Ab). A histological analysis of dorsal IPE lens structures after 17 days of Matrigel culture demonstrated the appearance of a defined outer cuboidal epithelial cell layer surrounding an inner lens fiber zone with visible parallel alignment of central lens fiber cells (Fig. 2B and Supplementary Fig. S2A) that stained positive for αA-crystallin (Fig. 2C). While earlier studies by Hayashi et al. demonstrated the generation of similar lens structures from dorsal IPE aggregates in Matrigel or collagen I culture supplemented with bFGF,4,5 bFGF does not seem to be a necessary requirement in our Matrigel culture system.
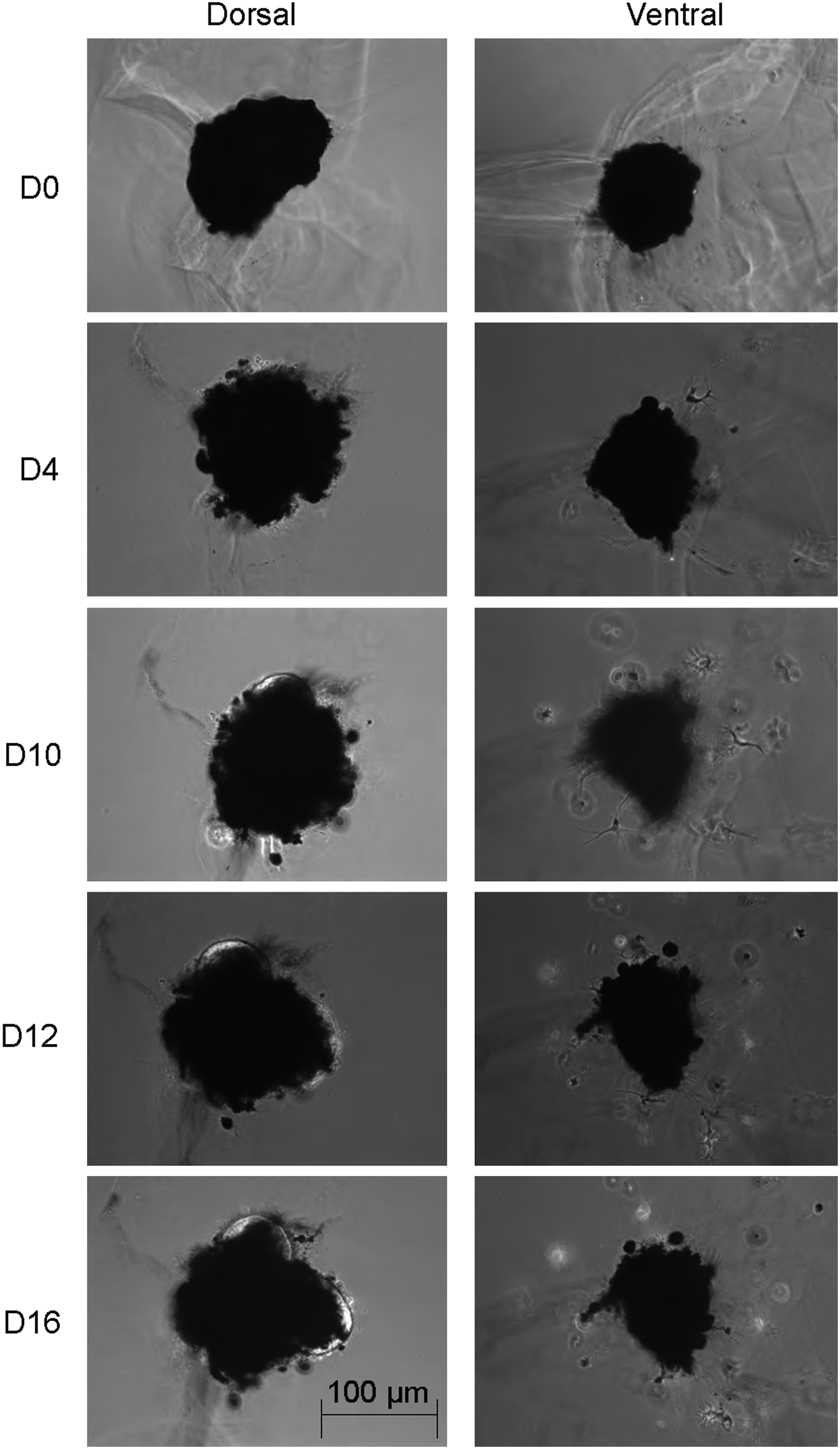
Morphological Changes of Newt iris pigment epithelial (IPE) Aggregates over Time. Phase-contrast images of whole newt IPE aggregates during the first 14 days of culture in Matrigel™. Images were taken using a 10× objective. Days (D) of culture are depicted on the left-hand side. Bar represents aggregate size in micrometer.
Lens Formation of Dorsal Newt IPE Aggregates in Matrigel
To test the functionality of in vitro generated lenses, dorsal and ventral IPE aggregates grown for 16 days in Matrigel were stained with vital dye CFDA-SE stained before re-implantation into the eye of a lentectomized newt (Supplementary Fig. S3Aa, b, c). Re-implantation of CFDA-SE-stained dorsal aggregates into the dorsal iris area resulted in complete lens formation besides the regenerated smaller host lens (Supplementary Fig. S3Ba, b). In contrast, CFDA-SE-stained ventral aggregates implanted into the dorsal iris area were distributed within the host pool of dorsal iris cells (Supplementary Fig. S3Ca, b, c).
Taken together, these data strongly support in vitro generation of functional lenses from dorsal IPE aggregates in our Matrigel culture system without the requirement of bFGF supplementation.
Matrigel promotes lentoid formation of MLE aggregates
Three-dimensional culture of mammalian primary LE cells has been used for almost 30 years to study lens-related healing and lens metabolism, with various outcomes regarding the generation of lens-like structures.6,9–12 To generate lens-like structures with consistent morphologies with regard to size, transparency, and participating cell types, we tested whether MLE cells were capable of forming lens-like structures after aggregation and culture in Matrigel similar to newt IPE cells.
To optimize MLE cell aggregation, different numbers of 5000, 35,000, 50,000, 100,000 and 500,000 cells were aggregated by centrifugation at 2000 g followed by overnight incubation at 37°C and transfer into low-growth-factor Matrigel. Optimal aggregation was achieved with 5000 to 50,000 cells (Fig. 3), while aggregates containing higher numbers of 100,000 to 500,000 cells were not stable and demonstrated partial dissociation of cells into the surrounding gel. The generated lens-like structures demonstrated similar characteristics to lentoids reported in previously established culture systems,6,9–12 including an overall rounded appearance with diffuse transparency throughout that increased mainly within the outer periphery over the 26 day culture period. In addition, different-size MLE aggregates of 5000, 35,000, and 50,000 cells demonstrated a volume reduction to proportional half of the original volume at the end of the culture period.
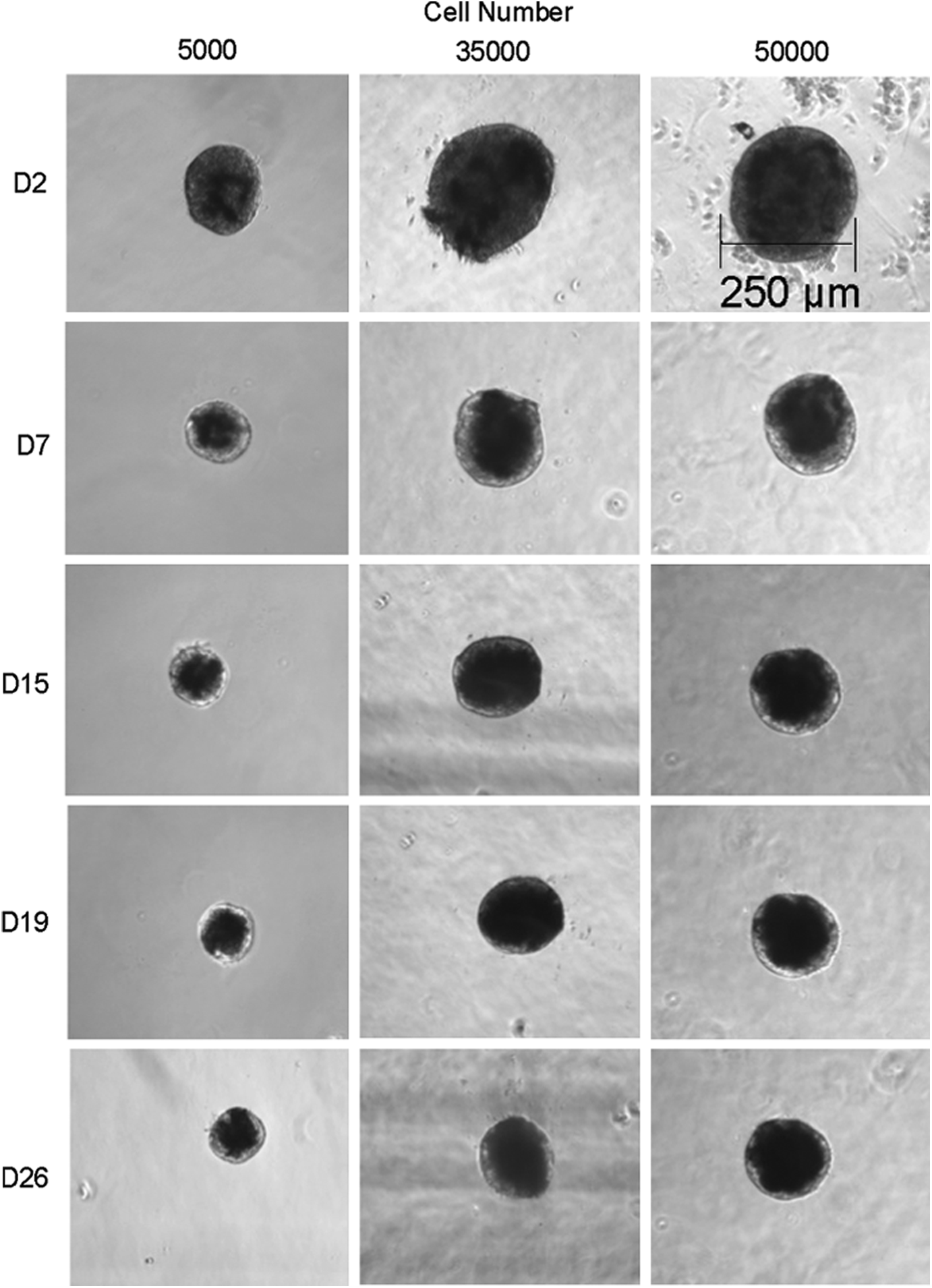
Morphology Changes of Different-Size Mouse Lens Epithelial (MLE) Aggregates over Time. Phase-contrast images of whole MLE aggregates containing 5000, 35,000, and 50,000 cells during the first 26 days of culture in Matrigel. Days (D) of culture are depicted on the left-hand side. Images were taken using a 10× objective. Bar represents aggregate size in micrometer.
MLE aggregates demonstrate lens-like central nuclear loss and establishment of a peripheral epithelial cell layer
To determine whether the time-dependent reduction in MLE aggregate volume can be linked to a lens fiber differentiation-associated nuclear loss,13–15 histology sections of aggregates were analyzed by phase-contrast microscopy in combination with Hoechst nuclear fluorescence staining (Fig. 4, see also Supplementary Fig. S4). At day 1 after plating in Matrigel, a diffuse cellular distribution could be observed in the aggregate center, while cells in the outer periphery appeared in sheet-like cell layers (Fig. 4a, b, c). At 1 week (Fig. 4d, e, f), aggregates demonstrated some volume loss and appeared more rounded in shape with cells and their corresponding nuclei occupying a more condensed space. In addition, the outer periphery demonstrated the formation of a well-defined border. After 2 weeks (Fig. 4g, h, i), aggregates demonstrated a further volume loss along with a partial central nuclear loss, while two to three sheets of cells in the outer periphery demonstrated intact nuclei. At 3 weeks (Fig. 4j, k, l), a further volume reduction and almost complete nuclear loss in the center of aggregates was observed with remaining intact nuclei in the outer peripheral cell layers. While previous studies have reported a partial nuclear loss in a diffuse fashion throughout the whole lentoid body, 16 the long-term maintenance of an epithelial type cell layer on the outer lentoid border has only been reported when the capsular bag is kept intact.17,18 To exclude the potential nuclear loss by apoptotic events, protein extracts from 0 day, 1, and 2 week aggregate culture were analyzed by caspase-3 immunoblotting. Immunoblotting analysis of aggregates from all culture periods lacked detection of active caspase 3, while the caspase 3 pro-form was present in all samples (data not shown). These data support the appearance of a nonapoptotic nuclear loss in the aggregate center that might be correlated with central lens fiber differentiation.13–15

MLE Aggregate Nuclear Loss over Time. Microscope images of Matrigel cultured MLE aggregate mid sections observed within 1 day
Increased MLE aggregate size and transparency in presence of bFGF
With regard to previous studies that demonstrated promotion of LE-dependent lens fiber and lentoid formation by bFGF,2,10,11 Matrigel culture of aggregates in regular medium was compared with culture in regular medium supplemented with 100 ng/mL bFGF (Fig. 5).
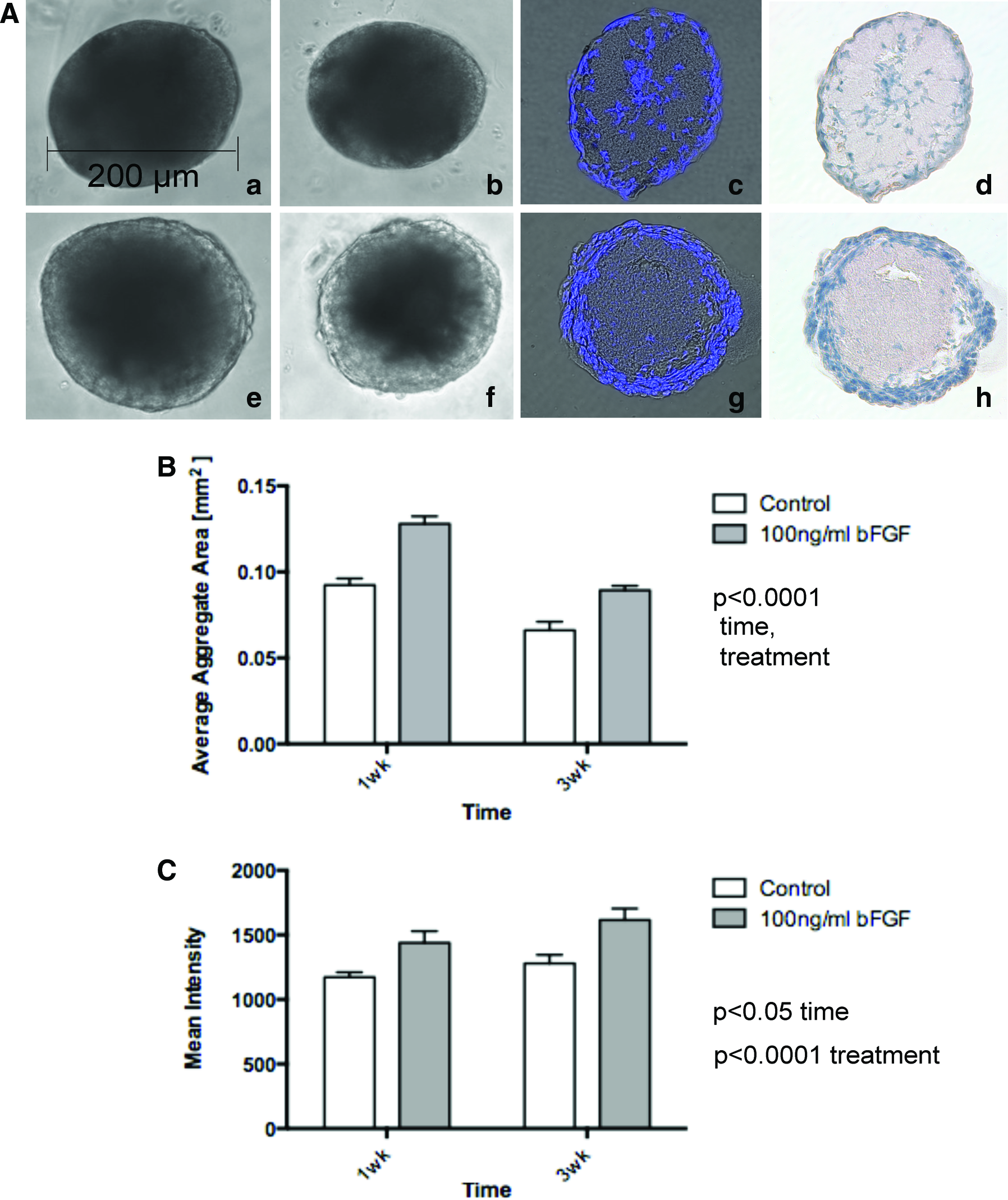
Time-Dependent Effect of Basic Fibroblast Growth Factor (bFGF) Treatment on MLE Aggregate Size and Transparency.
To measure detailed changes in aggregate size, aggregate areas of phase-contrast images depicted in Figure 5Aa, b, e, f were analyzed with NIH ImageJ 1.43s software by tracing the outline of aggregates (Fig. 5B). The average area of aggregates cultured for 1 week in the presence of bFGF was significantly higher with 0.127±0.004 mm2 compared with the 1-week control culture (0.092±0.003 mm2). In addition, bFGF-treated aggregates demonstrated a significant decrease in average aggregate area over time with 0.089±0.002 mm2 at 3 weeks that was in parallel with a corresponding decrease in the 3 week control aggregates (0.066±0.005 mm2, two-way ANOVA 1 week vs. 3 weeks p<0.0001 with regard to bFGF trea™ent and changes over time).
To determine transparency changes in the presence of bFGF treatment, phase-contrast images of aggregates depicted in Figure 5Aa, b, e, f were analyzed by densitometry using NIH ImageJ 1.43s by measuring increases in mean intensity (MI) representative for transparency increases (Fig. 5C). Corresponding to the main appearance of transparency on the outer aggregate edge, densitometry analysis included MI measurements from same-size area rectangles spanning from the outer aggregate edge toward the center. Aggregates cultured for 1 week in the presence of bFGF demonstrated a significantly higher MI with 1438.41±90.37 MI versus the control culture (1172.85±37.76 MI). At 3 weeks, aggregate mean intensities demonstrated a significant time-dependent increase in both bFGF (1615.35±89.51 MI) and control cultures (1279.34±65.74 MI) (two-way ANOVA 1 week vs. 3 weeks of bFGF treatment: p<0.0001, time: p<0.05).
In correlation with the observed bFGF-dependent increased average aggregate size and overall transparency, bFGF-treated aggregates demonstrated an increased appearance of nucleated sheet-like cell layers in the outer periphery compared with the nontreated control (Fig. 5Ag, h vs. 5Ac, d) (Supplementary Fig. S2B). These data suggest a bFGF-dependent stimulation of aggregate expansion through increased proliferation in the outer peripheral cell layers.
Changes in MLE aggregate βB1-crystallin expression in presence of bFGF
To determine whether the observed transparency in the MLE aggregate periphery is associated with increased crystallin production as an indicator of lens fiber formation,19,20 βB1-crystallin expression was analyzed by immunohistochemistry (Fig. 6). Due to the appearance of autofluorescence in aggregate paraffin histology sections, whole aggregates and whole newt lenses (control for antibody penetration within whole organ structures) were analyzed by confocal microscopy. At 1-week culture, βB1-crystallin fluorescence was mainly detected in the outer rim of lens aggregates that matched transparency areas within the corresponding transmission images. In contrast to control cultures, aggregates cultured in the presence of 100 ng/mL bFGF for 1 week demonstrated a more intense βB1-crystallin staining. At 3 weeks, both control and bFGF-treated cultures demonstrated spreading of βB1-crystallin toward the middle of the aggregates.
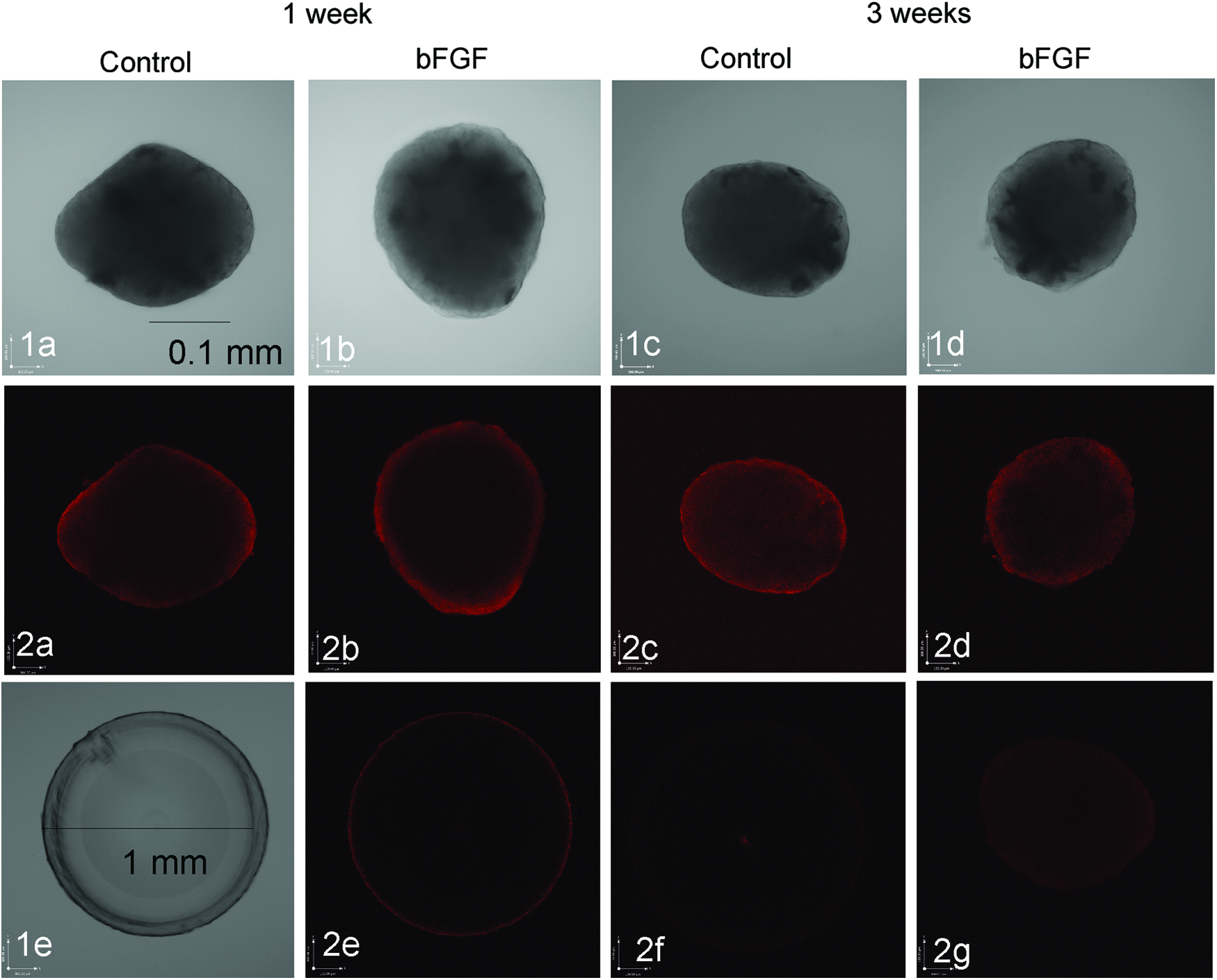
Time-Dependent Changes in MLE Aggregate βB1-Crystalllin Detection After bFGF Treatment. Confocal microscope images taken through 10 micrometer mid sections of whole aggregates at 20× objective
Due to the fact that whole newt lenses demonstrated βB1-crystallin staining only within the outer rim of the lens (Fig. 6, image 2e), it cannot be excluded that the used staining method lacks complete penetration of the βB1-crystallin antibody into the cell aggregates.
The formation of β-crystallin homo- or heterodimers and high-molecular-weight form oligomers consisting of β-crystallin family members, for example, βB1-, βB2-, βB3-,βA3/1-, βA2-, and βA4-crystallin were previously suggested to be essential for the close packing and stabilization of lens proteins and the formation of a clear lens. 21 To detect the bFGF-dependent changes in βB1-crystallin expression and composition over time, MLE aggregates were analyzed by Western blotting at different time points, including before plating (t0) and after 1 and 2 weeks of culture in Matrigel (Fig. 7). While βB1-crystallin in its monomeric form was not detected in any of the MLE aggregates, immunoblotting detected two bands at proportional 58 and 200 kDa corresponding to potential dimeric and multimeric forms of βB1-crystallin, respectively (Fig. 7A). Protein extracts from commercially available bovine β-crystallin, whole lens, (Fig. 7C), and lens fiber protein extracts (Fig. 7A) matched the βB1-crystallin detection in MLE cells and aggregates with regard to the 200 kDa band but also demonstrated the detection of other bands at 49, 29, and 20 kDa (Fig. 7C). These differences in βB1-crystallin composition of natural lens versus MLE aggregates suggest that βB1-crystallin might be assembled incorrectly in MLE aggregates.
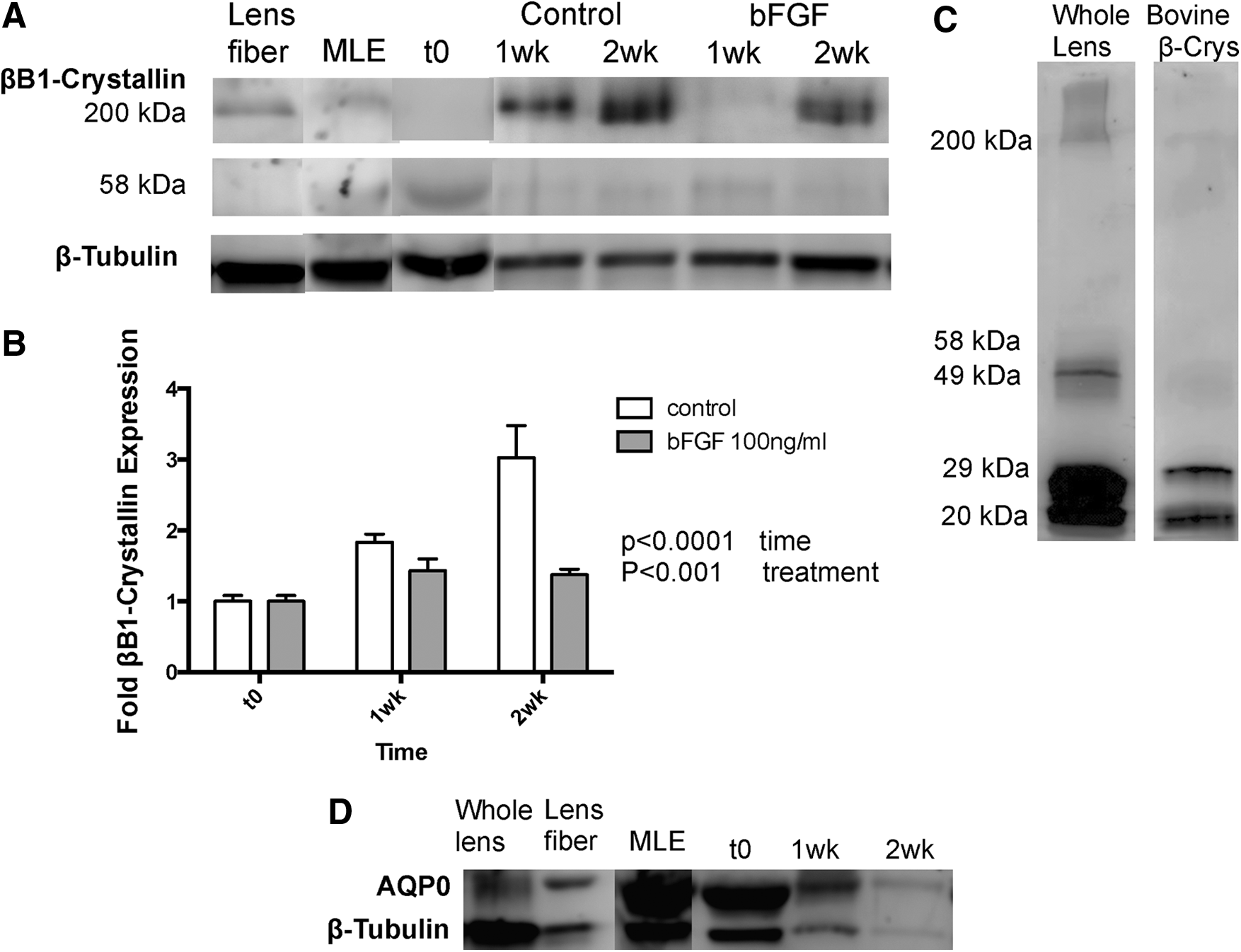
Time-Dependent Changes in MLE Aggregate Lens-Specific Markers After bFGF Treatment.
This is further supported by observations of βB1-crystallin expression in MLE aggregates over a 2-week time period (Fig. 7A, B). In contrast to βB1-crystallin expression in regular aggregate culture that demonstrated a significant 3.03±0.46-fold up-regulation of high-molecular-weight 200 kDa βB1-crystallin over a 2 week time period (p<0.0001, one-way ANOVA with Bonferroni post-test), bFGF treatment resulted in a marginal increase to 1.43±0.16-fold (p<0.05, one-way ANOVA with Bonferroni post-test) at 1 week that persisted at this level at 2 weeks (1.38±0.079) (two-way ANOVA p<0.0001 regarding bFGF treatment and p<0.001 regarding time-dependent changes). Expression of the βB1-crystallin 58 kDa dimeric form did not demonstrate any detectable significant changes in both control and bFGF-treated samples over the 2-week time period (data not shown). While the time-dependent increase in high-molecular-weight βB1-crystallin supports the process of lens fiber formation within MLE aggregates, the corresponding changes in the presence or absence of bFGF treatment further indicate that the βB1-crystallin assembly might be disorganized or incomplete within MLE aggregates.
In addition to βB1-crystallin as a marker for lens fiber differentiation, we also tested the expression of aquaporin 0 (AQP0) that is abundantly expressed in lens fibers.22,23 Western-blotting analysis demonstrated similar expression levels of AQP0 in protein extracts derived from whole lens, lens fiber, and MLE cells, and demonstrated no significant changes in MLE aggregates over the complete culture period of 2 weeks (Fig. 7D).
bFGF Treatment results in decreasing α-smooth muscle actin levels indicative of lens fiber differentiation
To determine the MLE aggregate expression of α-smooth muscle actin (αSMA) as a measure of lens fiber differentiation, protein extracts of MLE aggregates were analyzed by αSMA-specific immunoblotting (Fig. 8). Immunoblotting demonstrated detection of an αSMA specific band at 43 kDa that was detected in protein extracts from MLE cells and MLE aggregates but lacked detection in lens fiber extracts. Aggregates cultured for 1 week in regular medium demonstrated a significant down-regulation in αSMA expression to 0.82±0.05-fold compared with the t0 control (1.0±0.04-fold, p<0.05, one-way ANOVA with Bonferroni post-test) that remained down-regulated at 2 weeks (0.71±0.07-fold p<0.001). These results match our previous observations of decreasing αSMA expression during lens fiber differentiation in regenerating mouse lenses and support appearance of lens fiber differentiation in MLE aggregates rather than transdifferentiation.24,25 In the presence of bFGF expression, levels of αSMA demonstrated a further significant reduction within 1 week to 0.6±0.06-fold compared with the t0 control (p<0.001, one-way ANOVA with Bonferroni post-test) that persisted at this level at 2 weeks (0.57±0.03) (two-way ANOVA p<0.0001 regarding bFGF trea™ent and p<0.01 regarding time-dependent changes). The higher reduction of αSMA expression in the presence of bFGF matches the role of this growth factor in lens fiber formation and suggests increased appearance of lens fiber differentiation under these culture conditions.2,10,11

Time-Dependent Changes in MLE Aggregate αSMA Expression After bFGF Treatment. Upper panel: Western-blot immunodetection of αSMA, and β-Tubulin (loading control) in protein extracts from lens fibers, MLE cells, and MLE aggregates at the start of the experiment (t0), after 1 week (1wk) and 2 weeks (2wk) of culture in regular medium (control) and regular medium supplemented with 100 ng/mL bFGF. Lower Panel: Graph represents densitometric analysis of αSMA-detected bands in upper panel normalized against the β-Tubulin housekeeping control. At least three samples containing 6–12 aggregates/sample were analyzed per time point (t0, 1wk, and 2wk) in control and bFGF-treated cultures. Statistical analysis: Two-way ANOVA, p<0.05 was used as a criterion for statistical significance.
Discussion
In this study, we developed a novel method to generate lenses from dorsal newt IPE cells and, respectively, defined size lens-like structures from MLE cells in vitro using cell aggregation and culture in growth factor-reduced Matrigel. In line with species-related differences in the stemness of lens regenerating tissues, for example, newt IPE cells that originate from neural tube ectoderm versus MLE cells which originate from the lens placode-forming surface ectoderm, our experiments demonstrated differences in the lens-forming potential of both tissues and their responsiveness to bFGF.
With regard to the intrinsic lens-forming potential of dorsal newt IPE cells, our study suggests that the provision of a 3D growth matrix such as Matrigel is essential for the induction of lens formation. When cultured in a suspension culture, around 70% of dorsal aggregates lacked the capability to form a lens in vitro. Interestingly, the addition of 100 ng/mL bFGF to the regular L15 culture medium resulted in 100% lens formation within 7 to 10 days of culture (Supplementary Fig. S1A). These results suggest that the lack of growth matrix can be, to some part, substituted by the addition of growth factors. In contrast to the studies of Hayashi et al.4,5 that used 3D aggregate culture in the presence of bFGF, our study presented Matrigel culture system does not require bFGF supplementation and results in almost complete lens formation of dorsal IPE aggregates with the appearance of central lens fiber cells and a peripheral cuboidal epithelial cell layer while ventral aggregates lack lens formation under these conditions. In addition, similar results of dorsal IPE aggregates with lack of lens formation in ventral IPE aggregates were observed when Matrigel culture was supplemented with bFGF (Supplementary Fig. S1B).
One explanation for the reported differences in experimental model systems with similar outcomes might be based on chosen culture conditions. For instance, the group of Hayashi et al. used 6 day-old aggregates derived from freshly purified IPE cells without any collagen preculture, and the corresponding lens formation under bFGF supplementation was achieved within 28 to 30 days. 4 The 2-week collagen-based preculture of IPE cells before IPE cell aggregation in our study might precondition the IPE cells to form lens without the necessity to add any supplemental growth factor. Correspondingly, the partial submersion culture of aggregates by growth on top of collagen I or Matrigel-coated dishes according to Hayashi et al.4,5 might require the addition of bFGF to compensate for the lack of the surrounding stimulatory growth matrix.
In line with lens-like structures generated by other methods,6,9–12 the presented MLE aggregation method requires certain MLE cell intrinsic functions. For instance, when testing the required number of MLE cells for spontaneous formation of lens-like structures, we found that lentoid formation can be observed when plating a number of proportionally 500,000 cells dispersed in 100 μL Matrigel. These spontaneous lens-like structures were smaller or equal in size with centrifugation-derived aggregates containing a number of 5000 cells (Supplementary Fig. S5Aa, b). Correspondingly, centrifugation represents a means to increase the amount of cells that participate in the formation of lens-like structures by providing closer proximity of participating cells, but a certain self-driven lens formation potential of the participating cells is still required. For instance, a Matrigel culture of primary YC5 mouse IPE cell aggregates from passage 5 did not result in lens-like structure formation, thus supporting a lack of lens regenerative function of these cells in mice (Supplementary Fig. S6). According to our observations, the intrinsinc capability of MLE cells to form aggregates after centrifugation and overnight incubation decreases after passage number 15. One of the reasons for the loss of aggregation potential might be the ongoing transdifferentiation of MLE cells into myofibroblast-like cells (epithelial-to-mesenchymal transition, EMT) that has been observed in similar primary LE cell lines. 26 Besides the formation of lens-like structures from aggregates grown in low growth factor Matrigel, we also observed similar aggregate morphologies when aggregates were plated in growth factor free collagen gel (Supplementary Fig. S5Ba, b, c), supporting an intrinsic capability of MLE cells to form these organized structures in gel-like growth matrixes even without the addition of growth factors.
One of the observed characteristics of MLE aggregates was a decrease in aggregate size over time that was accompanied with an internal nonapoptotic nuclear loss. The observed nuclear loss along with an increase in βB1-crystallin and a decrease in α-SMA expression supports aggregate internal lens fiber differentiation that might lead to a tighter lens fiber cell packing and according to volume loss in our system.21,27
When MLE aggregates were cultured in the presence of lens fiber differentiation supporting growth factor bFGF,10,11,28–30 the aggregate structures demonstrated an initial increase in aggregate size at 1-week bFGF treatment that seemed to be based on the addition of outer peripheral cell layers. Despite the display of an increased transparency within the outer aggregate layers in parallel with βB1-crystallin production in this area, the outer periphery of bFGF treated aggregates seemed to lose its borderline shape integrity (Fig. 5A and Supplementary Fig. S2B). While previous studies have reported some beneficial effects of bFGF with regard to LE cell proliferation and lens fiber differentiation, the bFGF concentration seems to be highly relevant. For instance, Matrigel supplementation with 50 ng/mL bFGF in our system demonstrated the formation of lens-like structures similar to control aggregates (Supplementary Fig. S7). In a study by Azuma and Shearer that analyzed LE-dependent lentoid formation in regular Matrigel concentrations of 125–1000 ng/mL, FGF causes disruption of the typical rounded lentoid shape in association with the formation of elongated cellular processes that expanded over the original lentoid margin. 6 In another study by O'Connor and McAvoy that tested formation of lens-like structures in lens capsular explant cultures, the treatment with 100 ng/mL bFGF yielded more irregular structures in comparison to the use of eye inherent vitreous fluid. 18 Correspondingly, a combination of growth factors similar to those found in vitreous fluid might be beneficial for achieving ideal mammalian lens-like structures with the help of cell aggregation. Alternatively, a combination of 100 ng/mL bFGF in combination with 20 ng/mL Wnt-3a was recently demonstrated by the group of Yang et al. to yield clear lentoid bodies from human ES cells and might also be beneficial for induction of MLE-dependent lens formation. 3
Conclusion
In this study, we have successfully developed a novel in vitro culture system that uses cell aggregation and immersion of newt IPE or MLE cell aggregates into growth factor-reduced Matrigel for the generation of lenses or defined-size lens-like structures, respectively. Our study demonstrates that bFGF is not required for the induction of newt IPE aggregate-dependent lens formation in our Matrigel culture system. In contrast, Matrigel culture supplementation with bFGF seems to be beneficial for enhancement of features observed in MLE-derived lens-like structures such as stimulation of peripheral cell expansion in correlation with appearance of increased optical transparency and βB1-crystallin expression. In conclusion, the generation of reproducible newt IPE-derived lenses and mouse LE-derived lens-like structures by the established culture system might be beneficial for studies that test lens regenerative factors and require fast outcomes with reliable repetition.
Footnotes
Acknowledgments
This study was funded by NIH grants EY10540 and EY16707 to PAT.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.