
Abstract
The use of embryonic stem cells (ESCs) to regenerate distal lung epithelia damaged by injuries or diseases requires development of safe and efficient methodologies that direct ESC differentiation into transplantable distal lung epithelial progenitors. Time-consuming culture procedure and low differentiation efficiency are major problems that are associated with conventional differentiation approaches via embryoid body formation. The use of a growth factor cocktail or a lung-specific cell-conditioned medium to enrich definitive endoderm for efficient differentiation of mouse ESCs (mESC) into alveolar epithelial progenitor type II cells (ATIICs) has been reported, but not yet successful for generating a homogenous population of ATIICs for tissue regeneration purpose, and it remains unclear whether or not those mESC-derived ATIICs possess normal biological functions. Here, we report a novel method using a genetically modified mESC line harboring an ATIIC-specific neomycinR transgene in Rosa 26 locus. We showed that ATIICs can be efficiently differentiated from mESCs as early as day 7 by culturing them directly on Matrigel-coated plates in DMEM containing 15% knockout serum replacement. With this culture condition, the genetically modified mESCs can be selectively differentiated into a homogenous population (>99%) of ATIICs. Importantly, the mESC-derived ATIICs (mESC-ATIICs) exhibited typical lamellar bodies and expressed surfactant protein A, B, and C as normal control ATIICs. When cultured with an air–liquid–interface culture system in Small Airway Epithelial Cell Growth Medium, the mESC-ATIICs can be induced to secrete surfactant proteins after being treated with dibutyryl cAMP+dexamethasone. These mESC-ATIICs can synthesize and secrete surfactant lipid in response to secretagogue, demonstrating active surfactant metabolism in mESC-ATIICs as that seen in normal control ATIICs. In addition, we demonstrated that the selected mESC-ATIICs can be maintained on Matrigel-coated plates for at least 4 days with robust proliferative capacity. When cultured in DMEM medium containing 10% FBS, mESC-ATIICs spontaneously differentiated into alveolar epithelial type I cells. Collectively, these data demonstrate that the genetically modified mESCs can be selectively differentiated into a homogenous population of functional ATIICs, providing a reliable cell source to explore their therapeutic potential in lung tissue regeneration.
Introduction
A
Recently published data have demonstrated that ESCs can be differentiated into ATIICs via embryonic body (EB) formation5,6 or co-culture of EBs with pulmonary mesenchyme. 7 However, these procedures are not efficient, generating only a very small percentage of ESC-derived ATIICs. 8 Strategies using a growth factor cocktail or a lung-specific cell-conditioned medium9,10 to enrich definitive endoderm for efficient differentiation of ESCs into ATIICs have been developed, but not yet successful for generating a homogenous population of ATIICs. A mixed population of cell derivatives in the differentiated cultures of ESCs is not suitable for lung tissue regeneration. The remaining pluripotent cells in the differentiating cultures carry a significant risk of producing teratomas after transplantation in vivo. Hence, a major prerequisite for using ESCs therapeutically is to achieve a pure population of ESC-derived cells. Recently, an essentially pure population of ATIICs has been derived from genetically modified human ESCs (hESCs). 11 However, random genetic insertions may limit their therapeutic application. In addition, poor long-term retention/survival of engrafted hESC-derived ATIICs in injured lungs of SCID mice 12 indicates that the mouse lung may reject hESC-derived ATIICs or not provide an appropriate microenvironment for hESC-derived ATIICs to rebuild the progenitor cell pools after injury. Therefore, mouse ESCs (mESCs) should be used to explore the possible long-term therapeutic benefit provided by ESC-derived ATIICs in mouse injury models.
In this study, we generated a mESC line harboring an ATIIC-specific neomycinR (NEOR) transgene in Rosa 26 gene locus to avoid possible genetic mutations caused by random vector insertions. With the genetically modified mESCs, we developed a reliable differentiation procedure to efficiently generate a homogenous population of ATIICs and characterized their biological functions in vitro.
Materials and Methods
Construction of 3′hprt.mSPCP-NEOR.Rosa26 targeting vector
One 4.8 kb mouse SPC promoter (mSPCP) fragment was digested with AscI and BssHII, and cloned into AscI site of the 3′hprt insertion targeting vector (a gift from Dr. Allan Bradley, The Wellcome Trust Sanger Institute, Cambridge, United Kingdom). The NEOR cDNA-poly(A) fragment digested with AseI and EcoRI was then added into the engineered NdeI and EcoRI site downstream of mSPCP. To knock-in the mSPCP-NEOR transgene into Rosa 26 gene locus, one 5 kb DNA fragment homologous to Rosa 26 gene was subsequently cut with MluI and EcoRI, and cloned into AscI and EcoRI site of the targeting vector. The resulting 3′hprt.mSPCP-NEOR.Rosa 26 targeting vector (Fig. 1) was then linearized by NdeI before transfection.
Transfection and culture of mESCs
The mESCs with C57BL/6 genetic background were routinely maintained in mitomycin-treated mouse embryonic fibroblast (MEF) feeder cells on six-well plates using mESC culture medium. The medium contains 85% DMEM (Millipore), 15% FBS (HyClone), 10 mM HEPES, 1 mM sodium pyruvate, 1% nonessential amino acid, 2 mM L-glutamine, 100 μg/mL penicillin, 100 μg/mL streptomycin (Millipore), 1.52×104 M 1-thioglycerol (Sigma-Aldrich), and 1000 U/mL leukemia inhibitory factor (LIF; Millipore). Approximately 1×106 mESCs were re-suspended in 100 μL of supplemented mESC Nucleofector Solution (VAPH-1001; Lonza), mixed with 5 μg of the linearized targeting vector, and then transfected by using Nucleofector II as previously described.11,12 The transfected cells were then placed on gelatin-coated 10-cm plates in mESC culture medium. Puromycin (0.5 μg/mL; Sigma) was added the next day to select stably transfected mESCs. The transfected mESC colonies that had survived puromycin selection were picked up 6 days after transfection, and then maintained in mESC culture medium for the next study.
PCR and Southern blot analysis of site-specifically targeted mESC clones
PCR was performed using primer A and B (all primers used in the study were listed in Table 2) to screen Rosa 26-targeted mESC clones, which was carried out at 94°C for 3 min followed by 30 cycles at 94°C for 30 s, 60°C for 30 s, and 72°C for 1.5 min using AmpliTaq Gold DNA polymerase (AB Applied Biosystems). For Southern blot analysis, genomic DNA samples isolated from Rosa 26-targeted mESC clones (32, 33, and 34) were digested with BamHI, HindIII, and NheI, respectively, for Southern blot hybridization using a 425 bp NEOR gene fragment as a probe.
In vitro derivation and culture of mESC-derived ATIICs
To spontaneously differentiate mESCs into ATIICs, the dissociated mESCs were cultured on Matrigel-coated or Collagen IV-coated six-well plates at a density of 2×104 cells/cm2 in hESC differentiation medium (hDM), 11 LIF-free mESC culture medium (mESC differentiation medium [mDM]), or SR medium. The SR medium contains 85% DMEM, 15% KnockOut™ serum replacement (Gibco), 10 mM HEPES, 1 mM sodium pyruvate, 1% nonessential amino acid, 2 mM L-glutamine, 100 μg/mL penicillin, 100 μg/mL streptomycin, and 1.52×104 M 1-thioglycerol. From day 6, the differentiating cultures were either maintained in the original medium (hDM, mDM or SR medium) or transferred into a serum-free medium 10 for an additional 2, 4, or 6 days (Fig. 3A). The serum-free medium contained 75% Iscove's-modified Dulbecco's medium, 25% Ham's F12 medium (Gibco), 0.05% BSA, 2 mM glutamine, 0.5 mM ascorbic acid (Sigma-Aldrich), 4.5×104 M 1-thioglycerol, 50 U/mL penicillin, 50 mg/mL streptomycin, 50 mg/mL heparin sulfate salt (porcine intestinal mucosal; Sigma-Aldrich), 0.5× of both N2 and B27 (without retinoic acid) supplements (Invitrogen), and 50 ng/mL FGF2 (Sigma-Aldrich). To select the mESC-derived ATIICs (mESC-ATIICs), the mESC-32 cells were cultured on Matrigel-coated 6-cm plates in SR medium in the presence of G418 (25 μg/mL; GIBCO) for 7, 9, or 11 days. For spontaneous differentiation of mESC-ATIICs into the alveolar epithelial type I cells (ATICs), G418-selected mESC-ATIICs from day 11 as well as isolated mouse primary ATIICs (mATIICs) were cultured in six-well plates with DMEM containing 10% FBS for 8 days. 12 The mATIICs were isolated by using the procedure described by Corti et al. 13 To examine the capacity of mESC-ATIICs to proliferate in vitro, G418-selected mESC-ATIICs were cultured on Matrigel-coated six-well plates (2.0×104/well) in MEF-conditioned SR medium with or without 30 ng/mL of Recombinant keratinocyte growth factor (rhKGF; R&D Systems) for 4 days. 12
RT-PCR and QRT-PCR analysis
To evaluate the differentiation of mESCs into ATIICs, the total RNA samples were isolated from the differentiating cultures of mESCs on days 7, 9, and 11 by using RNA Bee (Tel-Test). RT-PCR was performed to analyze the expression of ATIIC-specific SPC with SPC forward primer and reverse primer by using OneStep RT-PCR Kit (Qiagen). To demonstrate the ability of mESC-ATIICs to differentiate into ATICs, the total RNA samples were isolated from the differentiating cultures of mESC-ATIICs and mATIICs on days 0, 2, 4, 6, and 8 to analyze expression levels of Aquaporin-5 (AQP5), Podoplanin (T1α) and SPC. QRT-PCR was carried out by using TaqMan One-Step RT-PCR Master Mix Kit (AB Applied Biosystems) 12 with the following primer pairs and probes: (i) AQP5 forward primer, AQP5 reverse primer, and AQP5 probe; (ii) T1α forward primer, T1α reverse primer, and T1α probe; (iii) SPC forward primer II, SPC reverse primer II, and SPC probe; and (iv) 18S forward primer, 18S reverse primer, and 18S probe.
Immunofluorescent staining
The G418-selected mESC-ATIICs were dissociated on days 7, 9, or 11, and seeded on poly-D-lysine-coated cover slips. After being cultured in SR medium overnight, the cells were washed with PBS and fixed in 4% paraformaldehyde at room temperature for 10 min. Cells were then permeabilized in 0.5% Triton X for 30 min. After being blocked in 5% goat serum for 2 h, mESC-ATIICs were stained with 1:200 diluted rabbit anti-human proSPC, 1:500 diluted rabbit anti-human proSPB, or 1:500 diluted rabbit anti-human SPA antibody (Chemicon) as previously described. 11 The surfactant protein expression was visualized with Alexa Fluor 488 conjugated goat anti-rabbit IgG (1:1000; Molecular Probes) with Draq5 (1:500; Biostatus) counterstaining.
Electron microscopy
The ultra-structural examination of G418-selected mESC-ATIICs was performed by Electron Microscopy Laboratory, Department of Pathology, University of Texas Medical School at Houston.
Secretion of surfactant from cultured mESC-ATIICs
G418-selected mESC-ATIICs on day 11 were trypsinized and then seeded back onto fresh Matrigel-coated 10-cm culture plates with SR medium containing 3 H-choline (1 mCi/plate; PerkinElmer) for 24 h. Cells were washed with PBS and cultured in fresh SR medium with or without secretagogue (TPA, 50 ng/mL; Sigma-Aldrich) for 2 h at 37°C. 14 The medium was collected and combined with rinses from cell wash. The 3 H-labeled phosphatidylcholine (PC) in the medium and cells were extracted and counted, respectively, as reported. 15 To examine surfactant protein secretion, the mESC-ATIICs were trypsinized on day 6 and then cultured by using air–liquid–interface culture system in Small Airway Epithelial Cell Growth Medium (SAGM™; Chemicon Millipore) containing G418, with or without dibutyryl cAMP (Bt2cAMP, 1 mM) and dexamethasone (Dex, 10−10 M) for 5 days. 16 The proteins were harvested from culture medium and analyzed by Western blot with mouse monoclonal antibody against mature SPB (abcam) and rabbit anti-mature-SPC (Seven Hills Bioreagents) by using the previously described procedure. 17
Statistical analysis
The data shown in Figure 5B and C were analyzed with Student's t-test (two tailed) using Microsoft Excel software. Results with p-values less than 0.05 (alpha level=0.05) were considered statistically significant.
Results
Site-specific modification of mESCs
To generate site-specifically modified mESC lines, the targeting vector, 3′hprt.mSPCP-NEOR.Rosa26, was generated as depicted in Figure 1. The NdeI was used to linearize the vector before transfection. Of ∼1.0×106 mESCs subjected to the transfection, 312 survived the puromycin selection. PCR using primer A and B (Fig. 1) was performed to identify Rosa 26-targeted mESC clones. Since the primer A is located within Rosa 26 gene region upstream of NdeI site and the primer B in the NEOR gene downstream of the NdeI site in the vector, the PCR should only detect the Rosa 26-targeted transgene, but not those randomly inserted transgene fragments. Of 312 stably transfected clones, 29 were found to be Rosa 26-targeted mESC clones. As shown in Figure 2A, the stably transfected mESC clones 32, 33, 34, and 39 contain one 1.35 kb targeted transgene fragment, indicating a site-specific targeting at Rosa 26 gene locus. To select random vector-integration-free mESC clones, Rosa 26-targeted mESC clones 32, 33, and 34 were analyzed by Southern blot hybridization using a 425 bp NEOR gene fragment as a probe. The probe can recognize a 6.4 kb-targeted BamHI fragment, a 7.2 kb-targeted HindIII fragment, a 5.7 kb-targeted NheI fragment (Fig. 1), and a randomly inserted targeting vector. Southern blot analysis demonstrated that all of the three Rosa 26-targeted mESC clones contain the targeted BamHI fragment, HindIII fragment, and NheI fragment (Fig. 2B). No randomly inserted vector DNA was detected in mESC clone 32 (mESC-32), demonstrating only a single copy of the inserted transgene in Rosa 26 gene locus. Therefore, the mESC-32 was selected for the next study.

3′hprt.mSPCP-NEOR.Rosa26 targeting vector. Schematic diagram of 3′hprt.mSPCP-NEOR.Rosa26 targeting vector (showing relevant positional information, not scaled proportionally based on sequence lengths). The NEOR transgene controlled by mATIIC-specific SPC promoter (mSPCP-NEOR) was cloned into 3′hprt targeting vector backbone, containing the puromycinR gene (PURO), the K14Agouti transgene (Ag), and an LoxP site (arrow). One 5.0 kb DNA fragment homologous to Rosa 26 gene was included in the vector for site-specific targeting by homologous recombination. To generate Rosa 26-targeted mESCs, NdeI was used to linearize the vector before transfection of mESCs. The primer A is located within Rosa 26 gene region upstream of the NdeI site and the primer B within the NEOR gene downstream of the NdeI site in the targeting vector. Thus, PCR can be performed by using primers A and B to identify Rosa 26-targeted mESC clones. mESC, mouse embryonic stem cell; ATIIC, alveolar epithelial progenitor type II cell; mATIIC, mouse primary ATIIC; NEOR, neomycinR; SPC, surfactant protein C.
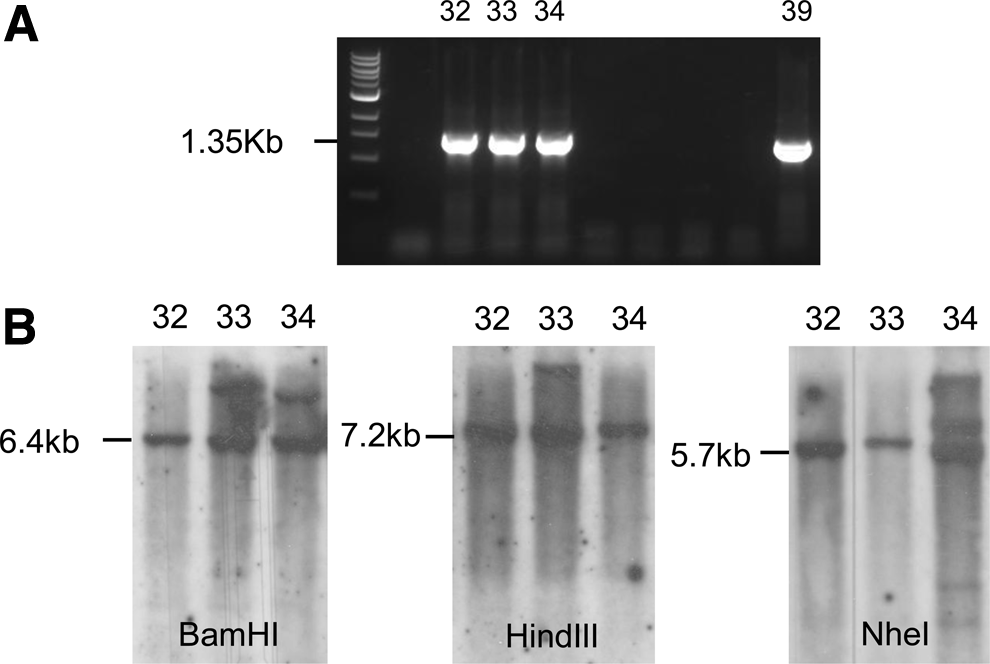
Identification of Rosa 26-targeted mESC clones.
Derivation and purification of mESC-ATIICs
To establish an optimal differentiation procedure for direct derivation of mESCs into ATIICs, mESCs were dissociated and seeded on either Matrilgel-coated or Collagen IV-coated six-well plates in hDM, mDM, or SR medium for the first 5 days. The differentiating cultures were then either maintained in the same differentiation medium or switched to the serum-free medium supplemented with 50 ng/mL FGF2 for an additional 2, 4, or 6 days (Fig. 3A). The differentiation of mESCs into ATIICs was evaluated by RT-PCR to analyze SPC expression in the differentiating cultures of mESCs. The expression of SPC RNA was detected in the differentiating cultures of mESCs in the Collagen IV-coated plates only when mDM was used for the first 5 days' differentiation (Fig. 3B). In comparison, significantly higher expression levels of SPC RNA were detected in the differentiating mESC cultures in Matrigel-coated plates as early as on day 7 when either SR medium or mDM was used to initiate the differentiation during the first 5 days (Fig. 3B). Supplements with 50 ng/mL FGF2 significantly enhanced the SPC expression in the differentiating cultures in Matrigel-coated plates on days 7 and 9 when mDM was used in the first 5 days. However, the expression level of SPC RNA declined dramatically on day 11. The FGF2-induced SPC expression was not observed in the differentiating cultures of mESCs in Matrigel-coated plates when SR medium was used for the first 5 days' differentiation. Interestingly, a high expression level of SPC in the differentiating mESC cultures in Matrigel-coated plates was maintained over time in SR medium without a supplement of FGF2 (Fig. 3B). Therefore, the differentiation procedure by culturing mESCs on Matrigel-coated plates in SR medium was used for the next study. Approximately 13.8% of cells in differentiating cultures of mESC-32 spontaneously differentiated into ATIICs on day 7, which was demonstrated by immunostaining for ATIIC-specific SPC expression (Table 1). The SPC-positive cells in the differentiating cultures increased to 15.4% on day 9 and 19.1% on day 11. The percentages of SPC-positive cells in the differentiating cultures of mESC-32 were comparable to those in the differentiating cultures of control mESCs. To isolate mESC-ATIICs, mESC-32 cells were subjected to spontaneous differentiation in the presence of 25 μg/mL of G418 for 7, 9, or 11 days. Since the SPC protein is specifically expressed by ATIICs, NEOR should be only expressed when mESC-32 cells are differentiated into ATIICs. Therefore, only ATIICs derived from mESC-32 can survive G418-selection. Immunostaining for SPC expression was carried out to determine the purity of selected mESC-ATIICs in the differentiating cultures. The number of SPC-positive cells was counted per 1000 cells based on DAPI staining on each plate. The results showed that more than 99% of cells were SPC positive in the differentiating cultures as early as on day 7 (Table 1), indicating that the G418 selection had efficiently eliminated the SPC-negative cell types derived from mESC-32 cells in the cultures. A similar percentage of SPC-positive cells was also present in the differentiating cultures on days 9 and 11. Interestingly, the number of SPC-positive cells in the G418-selected differentiating cultures significantly increased from 2.3×105/plate on day 7 to 4.8×105/plate on day 9 and to 1.4×106/plate on day 11 (Table 1), suggesting that G418-selected mESC-ATIICs continued to proliferate during the differentiation time period from day 7 to day 11.

In vitro differentiation of mESCs into ATIICs.
SPC+, mES cell-derived ATIICs; %, percentage of SPC-positive cells; #, the number of SPC+ cells in one 6-cm plate; mESC, mouse embryonic stem cell; ATIIC, alveolar epithelial progenitor type II cell; SPC, surfactant protein C.
Lamellar bodies and surfactant protein expression in mESC-ATIICs
ATIICs synthesize and secrete pulmonary surfactant, which are essential for maintaining homeostasis of alveoli. The surfactant lipids and proteins are stored in lamellar bodies, which are characteristic inclusion organelles in ATIICs. To examine whether these ATIIC-specific secretory organelles were present in the mESC-ATIICs, the ultra-structure of mESC-ATIICs was analyzed by transmission electron microscopy. The mESC-ATIICs exhibited typical lamellar bodies containing tightly packed multi-lamellar lipid membranes as observed in mATIICs (Fig. 4B). In addition, immunostaining was performed to examine the surfactant protein expression in mESC-ATIICs. As expected, the G418-selected mESC-ATIICs expressed surfactant proteins A, B, and C on day 7, 9, and 11 as mATIICs (Fig. 4A).
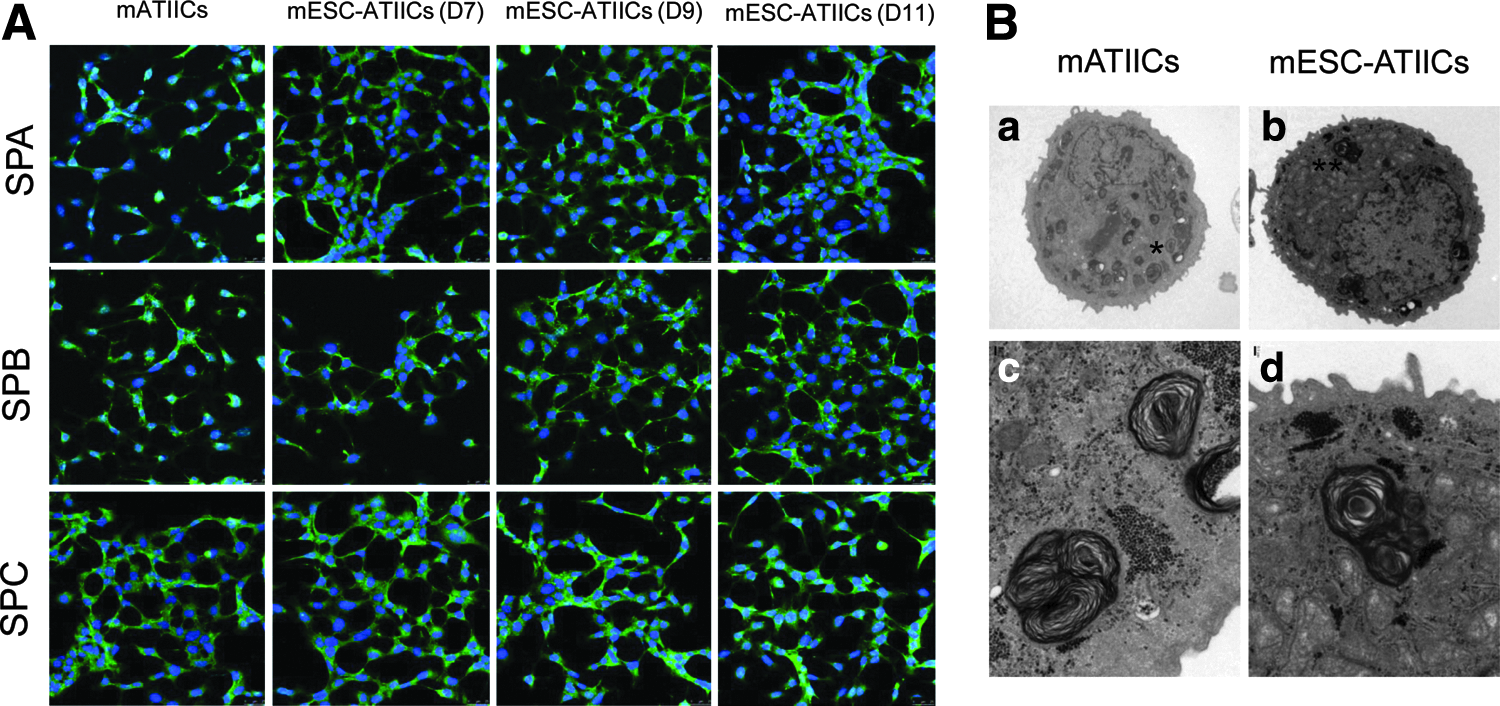
Surfactant protein expression and lamellar body formation in mESC-ATIICs.
In vitro proliferation and differentiation of mESC-ATIICs
One of the important biological functions of ATIICs is to serve as progenitor cells for repairing injured alveoli in response to injury. It is critical for developing cell-based therapies by using mESC-ATIICs to check whether the mESC-derived ATIICs possess the progenitor capacity. To demonstrate their ability to differentiate into ATICs, the mESC-ATIICs were subjected to spontaneous differentiation in DMEM containing 10% FBS for approximately 8 days. Total RNA was isolated from the differentiating cultures on days 0, 2, 4, 6, and 8 to analyze the expression levels of AQP5, T1α, and SPC. QRT-PCR demonstrated that the expression levels of AQP5 and T1α, the ATIC-specific markers,18–22 in the differentiating cultures of mESC-ATIICs significantly increased over time as those observed in the differentiating cultures of mATIICs (Fig. 5A). In support of the increased expression of the ATIC-specific markers, AQP5 and T1α, the expression levels of ATIIC-specific SPC dramatically decreased over time in the differentiating cultures (Fig. 5A). These data demonstrated the ability of mESC-ATIICs to spontaneously differentiate into ATICs in vitro. To examine their proliferative capacity, the G418-selected mESC-ATIICs were dissociated on day 11 and seeded back to Matrigel-coated six-well plates in MEF-conditioned SR medium. The rhKGF2, an ATIIC growth factor, 23 was added into some cultures to test whether rhKGF2 could enhance the proliferation of mESC-ATIICs. Many colonies were observed, and the number of mESC-ATIICs significantly increased from 2.0×104/well on day 0 to 3.5×104/well on day 2 and 4.9×104/well on day 4 in the cultures (Fig. 5B), suggesting that mESC-ATIICs proliferated in vitro. As expected, addition of rhKGF2 in the cultures promoted the proliferation of mESC-ATIICs, leading to a significantly greater number of cells (6.2×104/well on day 2 and 9.5×104/well on day 4) in the cultures compared with those in the culture in the absence of rhKGF2. Taken together, these data have demonstrated that G418-selected mESC-ATIICs cells possess the progenitor capacity of mATIICs.
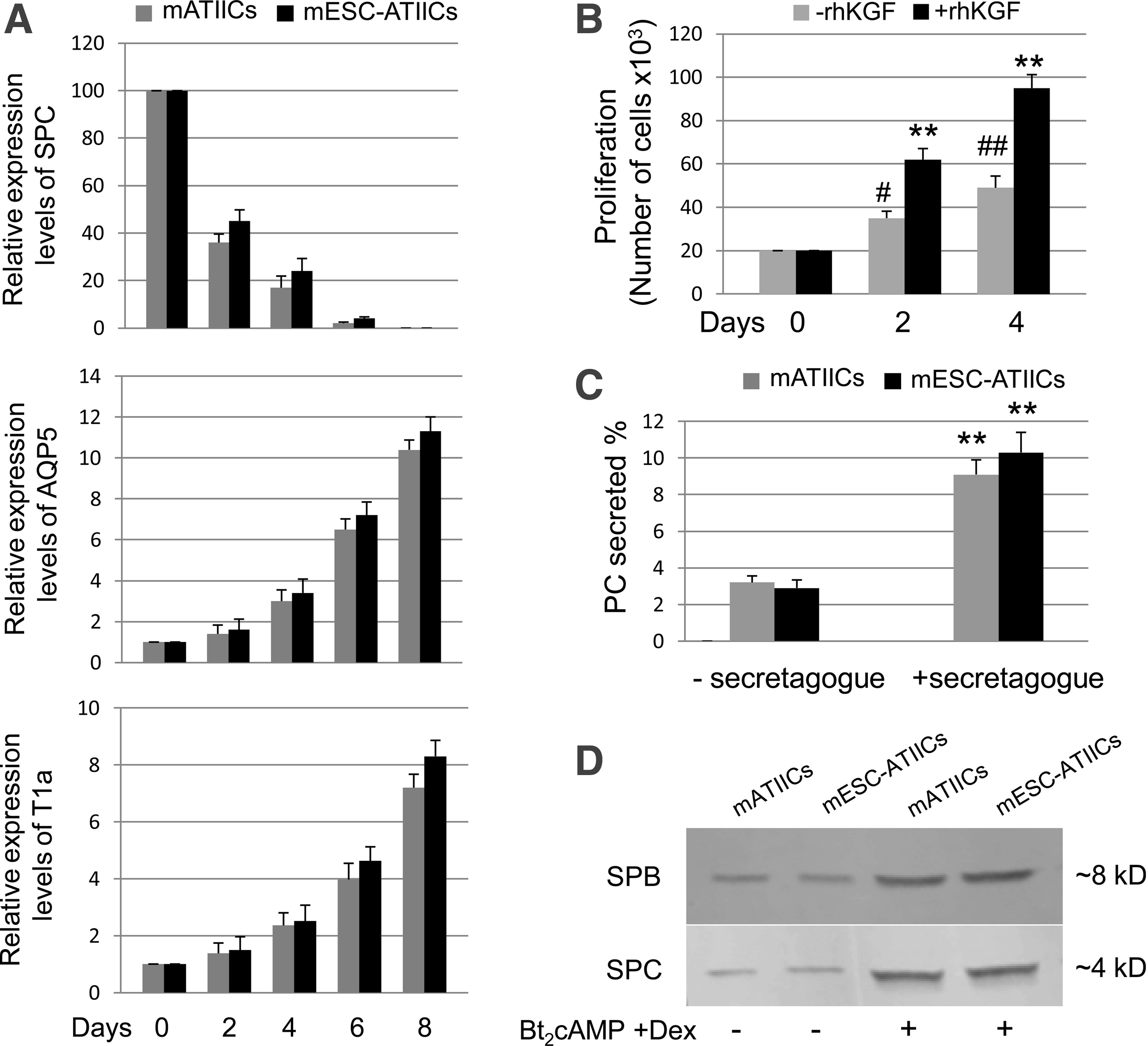
Biological functions of mESC-derived ATIICs.
Surfactant secretion from cultured mESC-ATIICs
It has been reported that cultured ATIICs recapitulate the expression and secretion of surfactant in a manner found in lungs. 16 To examine whether the mESC-ATIICs have acquired the ability, the cultured mESC-ATIICs were labeled with 3 H-choline to examine the surfactant lipid secretion in response to secretagogue. As seen in the cultures of mATIICs, the secretion of the 3 H-labeled PC (as a percent of total 3 H-labeled PC) from cultured mESC-ATIICs significantly increased in the presence of secretagogue (Fig. 5C). To further examine whether the expression and secretion of surfactant proteins from mESC-ATIICs can be regulated, the mESC-ATIICs were cultured with air–liquid–interface culture system in SAGM™ with or without Bt2cAMP and Dex for 5 days as described in the “Materials and methods” section. The protein samples harvested from culture medium were analyzed by Western blot. We found that control mATIICs and mESC-ATIICs synthesized and secreted SPC and SPB at very similar levels (Fig. 5D), whereas the secretion of SPC and SPB significantly increased in response to the treatment of Bt2cAMP and Dex. These data showed that the mESC-ATIICs synthesized and secreted surfactant in a regulated fashion, demonstrating active surfactant metabolism pathways in the mESC-ATIICs.
Discussion
Due to their numerous crucial physiological functions, ATIICs are a key target for developing therapeutic strategies to restore normal alveolar architecture and function that has been damaged by injury or disease. However, how to target ATIICs for correcting a mutated gene or expressing a therapeutic gene has remained elusive. Although bone marrow-derived stem cells (BMSCs) are being used to treat human heart failure and myocardial infarction, 24 recent investigations have demonstrated that either rare or no engraftment of BMSC-differentiated cells within injured lung epithelium.25–30 ESCs can be induced to differentiate into a wide range of different cell types in vitro,31–38 and, undoubtedly, can offer an attractive cell source for repair of injured/diseased alveoli.
Many methods have been reported for differentiating mESCs into ATIICs, yet all these approaches require complex or laborious culture conditions. Recently, Roszel et al. used Activin A and A549-conditioned medium to enrich definitive endoderm and differentiated mESCs seeded on Collagen IV-coated plates into ATIICs. 10 They found SPC expression on day 11 when the differentiation medium was supplemented with FGF2. We have previously reported that components of Matrigel facilitate differentiation of hESCs into ATIICs. 11 Here, we showed that significantly higher expression levels of SPC RNA were detected in the differentiating cultures of mESCs seeded on Matrigel-coated plates as early as day 7 in either mDM or SR medium, when compared with that in differentiating cultures of mESCs seeded on Collagen IV-coated plates. FGF2-induced SPC expression was observed in the differentiating cultures in Matrigel-coated plates on days 7 and 9 when mDM was used for the initial 5 days. However, this FGF2-induced SPC expression significantly declined on day 11. In comparison, a high expression level of SPC RNA in the differentiating cultures of mESCs in Matrigel-coated plates was well maintained in SR medium from day 7 to 11. Our results suggest that the choice of differentiation medium and coating matrix is key to efficient differentiation of mESCs into ATIICs. Characterization of protein components of Matrigel for identifying the particular proteins required to direct mESC differentiation into ATIICs can provide a significant insight into the mechanisms underlying mESC differentiation, and merits further investigation.
Expression of a fusion gene comprised a cell type-specific promoter and sequences encoding a resistance to neomycin, puromycin, or bleomycin in an ESC-derived cell lineage is a potential strategy to isolate tissue stem/progenitor cells derived from ESCs.11,39 However, the genetic abnormalities caused by genetic manipulation with random vector integrations are the major concern for the technique to be used in regenerative medicine. In this study, we used the site-specific targeting strategy to knock-in mSPCP-NEOR transgene into Rosa 26 gene locus of mESCs to avoid possible gene mutation caused by random vector insertions. With this single site-specifically modified mESC line, a homogenous population of mESC-ATIICs can be derived as early as on day 7 when mESCs were cultured on Matrigel-coated plates in SR medium. These mESC-ATIICs expressed SPA, SPB, and SPC with a characteristic ultra structure of normal ATIICs. The purity of derived ATIICs was well maintained in the presence of G418, as derived ATICs were eliminated immediately. It was observed in the study that these early-derived ATIICs continued to robustly proliferate in the differentiating cultures in the presence of G418, leading to a significantly increased number of mESC-ATIICs in differentiating culture (1.4×106 mESC-ATIICs/6-cm plate) on day 11. Since SPC expression has been observed in the more primitive lung precursor cells, 40 whether these early mESC-derived SPC positive cells on day 7 are the progenitor for ATIICs needs to be further characterized. However, this differentiation condition combined with use of the site-specifically modified mESCs can be scaled up to produce mESC-ATIICs in sufficient number to explore their therapeutic potential in alveolar regeneration.
It is critical to determine whether the mESC-derived ATIICs possess normal biological functions of control mATIICs for exploring their therapeutic potential. Previously reported data demonstrated the differentiation of mESCs into ATIICs by the evidence of surfactant protein expression and lamellar bodies in the derived cells. However, due to the lack of high purity of mESC-derived ATIICs for further characterization, it remains unclear whether or not those mESC-derived ATIICs possess normal biological functions. The site-specifically modified mESCs established in the present study can be selectively differentiated into a homogenous population of ATIICs in vitro, providing a sufficient number of mESC-ATIICs for us to characterize their biological functions. Our data demonstrated for the first time that the mESC-ATIICs possess progenitor capacity, and recapitulate the expression and secretion of surfactant in a manner found in the lungs. For example, they synthesized and secreted surfactant lipids, and can be induced to secrete surfactant proteins as control mATIICs. These biological functions are particularly important for ATIICs to maintain alveolar homeostasis. Thus, our results demonstrated that this novel strategy is reliable for producing a sufficient number of functional mESC-ATIICs to explore their therapeutic potential in lung tissue regeneration. The SPCP-NEOR transgene located in one known gene locus may still be a potential safety concern in practical application. “Clinical grade” ESC-derived ATIICs need to be generated for tissue regeneration in future.
In conclusion, we have successfully established an mESC line with a single copy of mSPCP-NEOR transgene in Rosa 26 gene locus. The genetically modified mESCs can be selectively differentiated into a homogenous population of functional ATIICs in a sufficient number when they were cultured on Matrigel-coated plates in SR medium. This novel strategy represents an important step toward exploring the therapeutic potential of mESC-derived ATIICs in lung tissue regeneration.
Footnotes
Acknowledgments
This work was supported by The Brown Foundation Institute of Molecular Medicine, University of Texas Medical School at Houston, and U.S. Public Health Service National Institutes Health Grants R21 HL102833-01(D.W). The authors thank Dr. Eva M. Zsigmond for providing the mESCs.
Disclosure Statement
No competing financial interests exist.