
Abstract
Neuronal signal transduction and communication in vivo is based on highly complex and dynamic networks among neurons expanding in a three-dimensional (3D) manner. Studies of cell–cell communication, synaptogenesis, and neural network plasticity constitute major research areas for understanding the involvement of neurons in neurodegenerative diseases, such as Huntington's, Alzheimer's, and Parkinson's disease, and in regenerative neural plasticity responses in situations, such as neurotrauma or stroke. Various cell culture systems constitute important experimental platforms to study neuronal functions in health and disease. A major downside of the existing cell culture systems is that the alienating planar cell environment leads to aberrant cell–cell contacts and network formation and increased reactivity of cell culture-contaminating glial cells. To mimic a suitable 3D environment for the growth and investigation of neuronal networks in vitro has posed an insurmountable challenge. Here, we report the development of a novel electrospun, polyurethane nanofiber-based 3D cell culture system for the in vitro support of neuronal networks, in which neurons can grow freely in all directions and form network structures more complex than any culture system has so far been able to support. In this 3D system, neurons extend processes from their cell bodies as a function of the nanofiber diameter. The nanofiber scaffold also minimizes the reactive state of contaminating glial cells.
Introduction
I
It was suggested that two-dimensional (2D) neuronal cultures benefit from the presence of co-cultured astrocytes. 7 Under normal conditions, astrocytes can release various factors that support the growth and development of neurons both in vivo and in vitro. 8 However, in situations such as neurotrauma or stroke, astrocytes become reactive, a condition that is defined by increased cell proliferation and cell process hypertrophy, upregulation of stress proteins such as glial fibrillary acidic protein (GFAP), and release of various factors and cytokines,9–11 which can be both supportive of and detrimental to neuronal survival. Astrocyte reactivity and hyperproliferation are two undesired hallmarks of 2D cell culture environments.1,12 In a 2D culture system, contaminating astrocytes release various stress-induced factors, which modify the neuronal metabolism, often in a highly detrimental way. 13
Synaptogenesis, that is, the formation of neuronal synapses, is the key plasticity and regeneration-promoting phenomenon during brain development and in the adulthood. 14 To study neuron–neuron communication and signal transduction in vitro, neurons need to be placed in a cell culture environment that allows synaptogenesis to occur. Despite extensive studies, it yet remains to be elucidated whether neurons in the culture are capable of generating synapses autonomously, that is, without influence from glial cells. There is strong evidence that glial cells in neuronal cultures enhance the number of functional synapses,15–17 promote synapse maturation,18,19 and release signals that affect synaptogenesis.18,20,21
Hence, the existing culture systems for neurons suffer from severe limitations, and moreover, it remains unknown how the topographical features of 3D systems, such as nanofiber coating and diameter, scaffold porosity, and astrocyte contamination, influence properties of cultured neuronal cells. We report here the development of a novel highly defined 3D culture system for neurons, which supports neurite outgrowth and 3D network development while minimizing the reactivity of contaminating astrocytes.
Materials and Methods
Nanofiber preparation
The solutions for electrospinning were prepared by mixing biocompatible polyether-based polyurethane (PU) resin in a mixture of tetrahydrofuran (THF) and N,N-dimethylformamide (DMF). The solutions were mixed for 24 h and transferred to a syringe with a cutoff metal cannula acting as a nozzle during electrospinning. Common for all the electrospinning process recipes used were a feeding rate of 2 mL/h, 18 kV applied to the metal cannula, and an 18-cm gap between the nozzle tip and the collector. The fibers were collected directly onto a specially designed plastic ring fixture consisting of two intercalating rings, which allowed the fibers to be retained in between the two ring structures. These fixtures were then used as inserts in a standard culture wells. To prepare scaffolds with specific fiber diameters, we developed several different recipes and selected three of them that rendered distinct fiber diameter distributions. The three fiber diameter distributions selected for evaluation as scaffolds for the culture were 2500±390, 1350±250, and 450±120 nm. The solution used to spin the 2500±390 nm fibers was 52.2 wt% THF, 34.8 wt% DMF, and 13 wt% PU, using a 19-G metal cannula. The solution used to spin the 1350±250 nm fibers was 53.4 wt% THF, 35.6 wt% DMF, and 11 wt% PU, using a 21-G metal cannula. The solution used to spin the 450±120 nm fibers was 54.6 wt% THF, 36.4 wt% DMF together with 9 wt% PU and 0.045 wt% of NaCl. The addition of NaCl increases the conductivity of the solution and was required when spinning the 450 nm fiber diameters to fabricate bead-free fibers.
Nanofiber coating
Nanofibers were sterilized with 70% ethanol, washed in dH2O, and incubated with poly‐
Neuronal cell cultures
All mice were housed in a barrier facility, and experiments were conducted according to protocols approved by the Ethics Committee of the University of Gothenburg. Pregnant mice of C57Bl6-129Sv-129Ola mixed genetic background at 16 days of gestation were sacrifized and embryos were decapitated. Hippocampi were dissected and freed from meninges, washed twice in cold HBSS, and incubated for 10 min at 37°C in 0.25% Trypsin solution. Trypsin was removed and Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (HyClone, Cat no. SH30071.03, batch no. ATE32026) and DNase was added. Mechanical dissociation was gently performed with a fire-polished Pasteur pipette. Cell suspension was passed through a 40-μm mesh and seeded at a density of 130,000 cells/cm2 for immunocytochemistry and 190,000 cells/cm2 for cell lysis experiments.
Scanning electron microscopy
Samples were imaged in a scanning electron microscope (SEM) (JSM6301F; Jeol) using an acceleration voltage and probe current of 6 kV and 7×10−11 A, respectively. Before inspection, the samples were sputter coated with ∼10 nm gold to avoid charging effects causing image distortion. The fiber diameters and pore size of the samples were then estimated from SEM images by using an image processing software (ImageJ). The average diameter was obtained by averaging >80 measurements of individual fibers. Histograms for the pore size distribution were assembled from data obtained from the SEM images of the fibers.
Confocal imaging
Neuronal cultures were fixed in 4% paraformaldehyde for 20 min before immunocytochemistry and mounting protocols had been applied. Image stacks in z-direction (z-distance between slices<0.25 μm) were acquired using a Leica confocal microscope (Leica TCS SP2). 3D z-stack images were rendered using Volocity software (Perkin Elmer) at the Centre for Cellular Imaging, University of Gothenburg, Sweden.
Immunocytochemistry
Neuronal cultures were fixed with 4% paraformaldehyde followed by permeabilization with 0.1% Triton X-100 in phosphate-buffered saline (PBS). Unspecific binding sites were blocked in PBS containing 2% normal donkey serum and 2% normal goat serum (blocking solution). After blocking, cells were incubated for 30 min with primary antibodies diluted in blocking solution, washed three times in PBS followed by secondary antibody incubation diluted in blocking solution. Primary antibodies used were rabbit anti-GFAP (Dako) and mouse anti-beta-III-tubulin (Covance). Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (Sigma). Secondary antibodies used were Alexa Fluor 488‐conjugated donkey anti‐rabbit, Alexa Fluor 594‐conjugated goat anti-rabbit, and goat anti‐mouse (Invitrogen). Images were taken with a Leica DMI 600B microscope.
5-Ethynyl-20-deoxyuridine incorporation
The uptake of 5-ethynyl-20-deoxyuridine (EdU) into the nuclei was detected using the click-iT kit (Invitrogen) according to the manufacturer's instructions. In brief, cell cultures were incubated with 10 μM EdU in the full culture medium for 3 h followed by 20 min cell fixation in 4% paraformaldehyde. Cells were permeabilized as described above, and EdU was detected using the click-iT reaction kit (Invitrogen).
Lactate dehydrogenase assay
Lactate dehydrogenase (LDH) assay was performed according to manufacturer's instructions (Takara Bio, Inc.). In brief, after dissection, neurons were plated in the medium comprising of the neurobasal medium (Invitrogen) supplemented with B27 (1×), penicillin (100 U/mL), and streptomycin (100 μg/mL). After 24 h, culture supernatant was collected, dead cells were removed by centrifugation (5 min, 300 rcf, 6°C), and the supernatant was used for the determination of LDH levels.
Protein extraction
Total protein of neuronal cultures from mice of C57Bl6-129Sv-129Ola mixed genetic background was obtained by adding protein lysis buffer to the cell culture plates for 2D or by submerging the Bioactive 3D inserts in an Eppendorf-tube containing lysis buffer for Bioactive3D cell cultures. The lysates were sonicated for 30 s with an amplitude of 14 μm before storing at −80°C until further usage. The lysis buffer composed of 20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.1% v/v Triton-X 100, one tablet protease inhibitor/10 mL lysis buffer (Roche), and one tablet phosphatase inhibitor/10 mL lysis buffer (Roche). The protein concentrations within the protein lysates were measured using BioRad DC protein assay (BioRad) and compared to BSA standards of known concentration.
Western blot analysis
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) was performed using Any-kD gels (Bio-Rad) with Tris-glycine running buffer (Bio-Rad). Blotting was conducted on PVDF membranes (Immobilon; Millipore) with 100 V per blot for 1.5 h. Transfer buffer composed of Tris-base (2.9 g/L), glycine (14.4 g/L), and MeOH (10% v/v) in dH2O. A total amount of 15 μg of lysate per lane was loaded. Unspecific binding was blocked using 3% BSA in Tris-buffered saline containing 0.1% Tween for 1 h at room temperature. Primary antibody incubation was performed overnight at 4°C followed by incubation with secondary horseradish peroxidase (HRP)-linked anti-rabbit or anti-mouse antibodies (Cell Signaling) for 45 min at room temperature. Secondary HRP antibodies were detected using chemiluminescence detection solution (Luminogen PS-3; GE Healthcare) and a LAS-3000 luminescent image analyser (Fujifilm), and the signal transformed into arbitrary units using MulitGauge software (Fuji). For normalization, beta actin signal was used. The following antibodies were used mouse anti-GFAP (Millipore), goat-anti MCM2 (Santa Cruz), mouse anti-glutamine synthetase (GS; Millipore), and rabbit anti-synaptotagmin (Abcam).
Statistical analysis
All results are presented as mean±standard error of mean. Data containing more than two groups were analyzed by two-way ANOVA, followed by Bonferroni post hoc comparisons, using GraphPad Prism 4.0 software. Significance was set at p<0.05. Data describing the percentage of EdU-positive cells and neurite extension in z-direction were analyzed by two-tailed Student's t-test.
Results
Neurons in the central nervous system have the ability to grow in all three dimensions, a property lost in 2D cell cultures due to the planar surface. We hypothesized that a nanofiber-based 3D cell culture system with highly defined and optimized characteristics, that is, biological coating, fiber diameter, and scaffold pore size, could restore the ability of neurons to extend neurites in the z-direction and allow such neurons to form complex 3D networks in the culture. Moreover, we hypothesized that such 3D cell culture system would minimize the reactive phenotype of contaminating astrocytes and thus bring another highly desirable benefit to neuronal cell culture system, namely reduced proliferation of contaminating astrocytes and release of undesired cell stress-induced factors by these cells.
PDL-coated PU nanofiber scaffolds are biocompatible with hippocampal neurons in vitro
PU is a class of elastomers that has been suggested to be biocompatible and could be used in soft tissue engineering. 22 Therefore, we selected a polyether-based PU as the 3D scaffold base material due to its suitable elastic properties and biocompatibility. We evaluated the viability of neurons on PDL-coated electrospun PU nanofiber scaffolds of various diameters and compared these scaffolds with PDL-coated (2D) Permanox plastic surfaces (Nunc). After 24 h, LDH survival assays were performed on neurons grown on coated 2D surfaces and on coated PU nanofiber scaffolds composed of nanofibers with 450, 1350 and 2500 nm in diameter. There were no differences in cell survival detected between coated 2D surfaces and nanofibers of 450, 1350 and 2500 nm diameter (Fig. 1A). The nanofiber scaffolds composed of a porosity (air to nanofiber volume ratio) of 63%±3%, 66%±5% and 68%±4% for nanofiber scaffolds with 450, 1350 and 2500 nm fiber diameter, respectively (Fig. 1B). The respective pore size distribution for the different nanofiber preparations indicates that productions of larger fiber diameter scaffolds resulted in increased pore size with the 2500 nm nanofiber preparations containing the largest pores with an area of 50 μm2 and above (Fig. 1C). Within the 450 nm nanofiber scaffolds, the majority of pores were 30 μm2 and below.

Nanofiber scaffolds for three-dimensional (3D) neuronal cell cultures.
Nanofiber diameter affects neurite outgrowth
Neurons in the living brain (in vivo) have the ability to form complex 3D networks of neuronal processes. To mimic this effect in the culture, neurons must be placed in culture conditions that promote and support the outgrowth of neurites. Therefore, we compared the effect of the 2D and 3D cell culture systems on the neurite extension of hippocampal neurons after 2 days in culture. We found that 51%±5% of neurons extended neurites when cultured on the 450 nm nanofibers compared to only 23%±12% and 23%±9% of neurons with neurites cultured on the 1350 or 2500 nm nanofibers, respectively (Fig. 1D). These results indicate neurite outgrowth to be a function of the nanofiber diameter and the pore size within the nanofiber scaffolds. Results obtained from the neurite outgrowth experiments demonstrated that 450 nm nanofibers best promoted neurite outgrowth and therefore, all following experiments were conducted on the 450 nm nanofiber scaffolds.
The nanofiber scaffolds support the formation of 3D neuronal networks
To test the hypothesis that 3D nanofiber scaffolds support the formation of complex 3D neuronal networks and allow neurons to extend neurites into the z-direction, that is, into the nanofiber scaffolds, we used confocal image analyses of neurons labeled for the neuron-specific cytoskeletal protein beta-III-tubulin. Neurons grown in the 3D cell culture were able to extend their neurites through the entire thickness of the nanofiber scaffold, in contrast to 2D cultures that due to the lack of a support structure were not able to extend any neurites into the z-direction (Fig. 2A). The maximal occupied space in the z-direction for 2D grown neurons was at the cell soma, with an average of 10 μm. The 3D grown neurons extended their neurites up to 25 μm in the z-direction (Fig. 2B–D). The ability of neurons to spread out and extend their neurites in the z-direction within the 450 nm fiber diameter scaffolds allowed these neurons to establish complex networks with a multitude of cell–cell contacts in the x-, y- and z-direction.
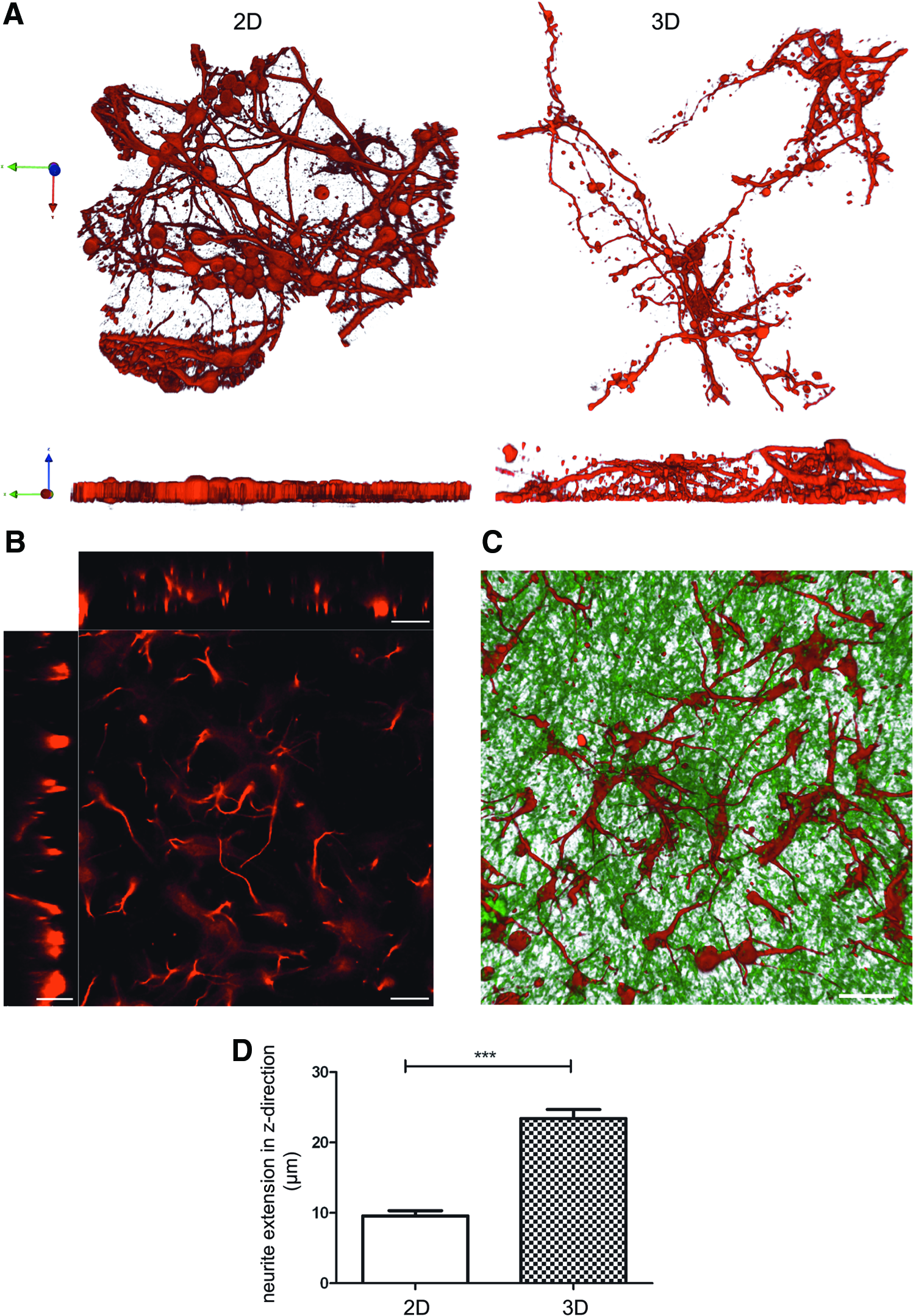
Morphology of the beta-3-tubulin cytoskeleton and neurite extension of hippocampal neurons cultured for 7 days in 2D and 3D revealed by 3D reconstruction of confocal images.
Contaminating astrocytes in 3D neuronal cultures are less reactive
Western blot analysis demonstrates that despite the presence of a comparable number of contaminating astrocytes in 2D and 3D, as determined by the presence of the astrocyte marker GS (Fig. 3A), the contaminating astrocytes in 3D express lower levels of the reactive astrocyte marker GFAP compared to astrocytes in the 2D neuronal cultures (Fig. 3B). This finding was further strengthened by the immunocytochemical analysis, which revealed a confluent monolayer of GFAP-positive astrocytes in 2D but not in 3D neuronal cultures (Fig. 3E).

Western blot and immunocytochemistry analysis of astrocyte contamination, reactivity, and proliferation in 2D and 3D neuronal cultures.
To further evaluate the astrocyte reactivity in neuronal cultures, we investigated cell proliferation by labeling dividing astrocytes in the 2D and 3D neuronal cultures by using an exogenous proliferation marker EdU. EdU, a uridine analog, is incorporated into the DNA of proliferating cells. Since neurons in culture do not proliferate, 23 only the proliferating fraction of the contaminating astrocytes was labeled in the neuronal cultures. Compared to 2D cultures, 3D neuronal cultures contained a smaller fraction of proliferating astrocytes (Fig. 3C). Western blot analysis for the proliferation marker MCM2 further confirmed reduced proliferation in neuronal cultures after 7 days in culture (Fig. 3A). Together these findings indicate that the contaminating astrocytes in 3D neuronal cultures are less reactive than in 2D neuronal cultures.
Neurons grown in 3D scaffolds exhibit synaptogenic potential
To test whether hippocampal neurons grown in 3D were capable of synaptogenesis, we investigated the expression of the presynaptic marker synaptotagmin in 2D and 3D culture systems. Western blot analysis revealed that synaptotagmin protein expression in neurons grown in 3D was comparable to that in 2D (Fig. 3D), indicating that the synaptogenesis process was not negatively influenced by the less reactive astrocytes in the 3D system.
Discussion
With the aim to mimic the extracellular environment and to provide structural support for cells in vitro, a number of 3D cell culture systems have been developed recently. The majority of these systems are based on hydrogen gels, 24 alginate and collagen gels,25,26 or electrospun polymer fibers,27,28 which are often used in combination with cell lines and neural progenitor cells.25,27,29 To date, it has not been investigated whether electropsun PU nanofiber scaffolds are suitable for primary neuronal cell cultures and whether neurite outgrowth and network formation are influenced by fiber diameter and scaffold porosity. Furthermore, a crucial factor influencing neuronal cell cultures, the reactive state of contaminating astrocytes, has not been thoroughly tested in these models. In the present study, we designed and utilized a bioactively coated electrospun PU nanofiber scaffold of specific nanofiber diameter for the neuronal cell culture. PU, a bioinert material, has been widely used in the biomedical field for many years, and its suitable biocompatibility has been recognized. The use of PU nanofiber scaffolds in our application shows excellent biocompatibility for neurons. Our results demonstrate that the 3D nanofiber scaffolds can maintain healthy neurons and support the outgrowth of neurites and formation of neuronal networks. It has been reported that pore size and structure can affect cell attachment and migration in vitro.30,31 Here, we demonstrate that, of the range of nanofiber diameters tested, the 450 nm nanofibers combined with small scaffold porosity promote neurite outgrowth much better than larger 2500 or 1350 nm nanofibers. This indicates that the neurite outgrowth is a function of nanofiber diameter and pore size. Furthermore, our nanofiber scaffolds support the extension of neurites into the z-direction of the scaffolds, allowing for complex neuronal networks to form, a property lost in other cell culture systems. Contaminating astrocytes, which frequently occur in neuronal cell cultures, showed reduced proliferation and decreased signs of cellular stress in the nanofiber scaffolds. We further demonstrate that a crucial attribute of neuronal cell cultures, the ability to generate synapses, was not negatively affected by our 3D nanofiber scaffolds.
Conclusion
We have achieved successful formation of complex 3D neuronal structures within a bioactively coated nanofiber network of highly defined parameters. Nanofiber diameter and scaffold pore size prove to be essential for neurite outgrowth. Neurons cultured within 3D scaffolds were capable of extending neurites in all three dimensions within the nanofiber scaffolds. Importantly, in our 3D culture system, contaminating astrocytes that naturally occur in neuronal cultures, were less proliferative and showed reduced expression of the astrocyte stress marker protein GFAP, which had been suggested to be a biomarker of neurotoxicity. 31 Thus, our 3D cell culture system for neurons provides a highly useful platform for the growth of and experimentation on live neurons.
Footnotes
Acknowledgments
This work was supported by the Swedish Medical Research Council (11548), ALF Gothenburg (11392), AFA Research Foundation, Söderbergs Foundations, Sten A. Olsson Foundation for Research and Culture, Hjärnfonden, Hagströmer's Foundation Millennium, Amlöv's Foundation, E. Jacobson's Donation Fund, VINNOVA Health Program, the Swedish Stroke Foundation, the Swedish Society of Medicine, the Free Mason Foundation, Chalmers University of Technology, NanoNet COST Action (BM1002), EU FP 7 Program EduGlia (237956), EU FP 7 Program TargetBraIn (279017).
Disclosure Statement
No competing financial interests exist.