
Abstract
Hydrogels can promote desirable cellular phenotype by mimicking tissue-like stiffness or serving as a gene delivery depot. However, nonviral gene delivery inside three-dimensional (3D) hydrogels remains a great challenge, and increasing hydrogel stiffness generally results in further decrease in gene delivery efficiency. Here we report a method to enhance nonviral gene delivery efficiency inside 3D hydrogels across a broad range of stiffness using sacrificial microfibers for co-releasing cells and polymeric nanoparticles (NPs). We fabricated hydrolytically degradable alginate as sacrificial microfibers, and optimized the degradation profile of alginate by varying the degree of oxidization. Scanning electron microscopy confirmed degradation of alginate microfibers inside hydrogels, leaving behind microchannel-like structures within 3D hydrogels. Sacrificial microfibers also serve as a delivery vehicle for co-releasing encapsulated cells and NPs, allowing cell attachment and spreading within the microchannel surface upon microfiber degradation. To examine the effects of sacrificial microfibers on nonviral gene delivery inside 3D hydrogels, alginate microfibers containing human embryonic kidney 293 cells and polymeric NPs were encapsulated within 3D hydrogel scaffolds with varying stiffness (9, 58, and 197 kPa). Compared with cells encapsulated in bulk hydrogels, we observed up to 15-fold increase in gene delivery efficiency using sacrificial microfibers, and gene delivery efficiency increased as hydrogel stiffness increased. The platform reported herein provides a strategy for enhancing nonviral gene delivery inside 3D hydrogels across a broad range of stiffness, and may aid tissue regeneration by engaging both mechanotransduction and nonviral gene delivery.
Introduction
H
The goal of the present study is to enhance the nonviral gene delivery efficiency inside hydrogels across a broad range of stiffness using biodegradable polymeric NPs. Specifically, we aim to use hydrolytically degradable alginate microfibers as sacrificial porogens to introduce void spaces inside 3D hydrogels, while simultaneously releasing cells and NPs inside macropores (Fig. 1). We hypothesize that transfection efficiency inside such hydrogels will increase as the bulk hydrogel stiffness increases, as the released cells will sense the bulk hydrogels more as a substrate. To determine microfiber compositions with optimal degradation profile, hydrolytically degradable alginate with varying degrees of oxidization was synthesized. Human embryonic kidney (HEK) 293 cells and polymeric NPs were co-encapsulated inside alginate microfibers with an optimized degradation profile, and then embedded within 3D bulk hydrogels. To fabricate bulk hydrogels with a broad range of stiffness, poly(ethylene glycol)-dimethacrylate with varying concentrations was used, and gelatin-methacrylate (Gelatin-MA) was included at a constant concentration to support cell adhesion. Outcomes were examined using scanning electron microscopy (SEM), fluorescence microscopy, mechanical testing, and live/dead staining. Quantitative luciferase assay was used to determine the transfection efficiency inside microfiber-containing hydrogels across a broad range of stiffness, and cells directly encapsulated inside 3D bulk hydrogels without sacrificial microfibers were included as controls.
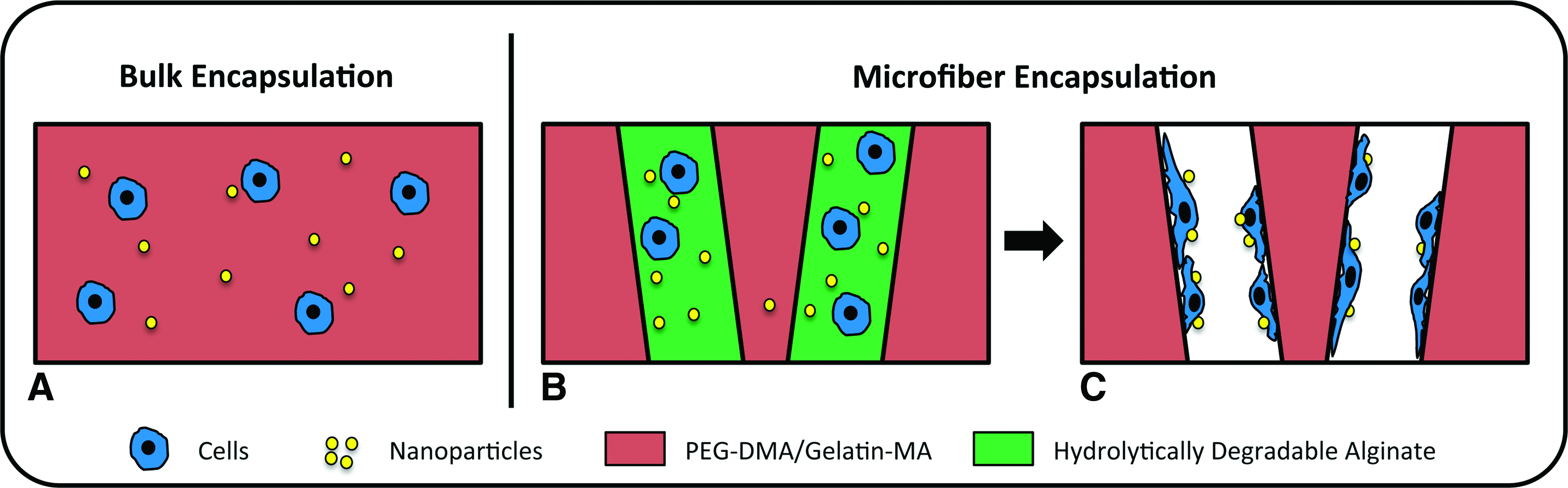
Schematic of encapsulating cells and nanoparticles (NPs) within bulk hydrogels
Materials and Methods
All materials were supplied by Sigma-Aldrich (St. Louis, MO) unless otherwise stated.
Synthesis of hydrolytically degradable alginate
Hydrolytically degradable alginate was prepared by oxidizing the diol linkages within the uronic acid monomers as previously described. 21 Alginate (MW=80–100 kDa) was dissolved to a final concentration of 1% (w/v) in distilled water. Sufficient sodium periodate was added to the stirring alginate solution to oxidize 3%–5% of the uronic acid monomers per alginate chain. The reaction was allowed to proceed overnight at room temperature, protected from light, and was quenched by the addition of an equimolar amount of ethylene glycol. The oxidized alginate was purified by dialyzing against distilled water for 2 days and lyophilized.
Synthesis of photocrosslinkable PEG and gelatin
Polyethylene glycol-dimethacrylate (PEG-DMA) was synthesized by reacting methacryloyl chloride with PEG diol (4.6 kDa), catalyzed by potassium carbonate and potassium iodide in dichloromethane overnight. The synthesized polymer was purified by dialyzing against DI water with 1-kDa molecular weight cutoff cellulose dialysis tubing (Fisher Scientific, San Jose, CA) for 2 days. The final polymer was lyophilized before use. Gelatin-MA was synthesized as previously described. 22 Briefly, type B gelatin was reacted with methacrylic anhydride under stirring at 50°C for 3 h. Gelatin-MA was extracted in acetone and purified by dialysis with 1-kDa molecular weight cutoff cellulose dialysis tubing (Fisher Scientific) for 2 days.
Synthesis of poly(β-amino ester)
Poly(β-amino ester)s (PBAEs) were synthesized as described previously.23,24 Briefly, hexanediol diacrylate (D) was reacted with 5-amino-1-pentanol 25 (Alfa Aesar, Ward Hill, MA) overnight at 90°C to produce acrylated terminated PBAE (D32). D32 was then reacted with excess tetraethleneglycoldiamine (122; Molecular Biosciences, Boulder, CO) at room temperature, and the resulting polymer (D32-122) was collected by extraction in ether. D32-122 was dissolved in anhydrous dimethyl sulfoxide (DMSO; Fisher Scientific) and stored at −20°C.
Synthesis of fluorescently labeled polymers
Fluorescently labeled gelatin was prepared by dissolving Gelatin-MA to a concentration of 7.5 mg/mL in distilled water. Ten microliters of a 50-mM solution of 5-caboxy-tetramethylrhodamine N-succinimidyl ester in DMSO was added, and the reaction was allowed to proceed overnight at 4°C, protected from light. The rhodamine-labeled methacrylated gelatin was dialyzed against distilled water and lyophilized.
Fluorescently labeled alginate was prepared based on a previously published procedure for conjugating peptides to alginate via carbodiimide chemistry. 26 Alginate was dissolved to 1% (w/v) in a 0.1-M MES and 0.3 M sodium chloride buffer solution at pH 6.5. N-hydroxysuccinimide (NHS) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) were added to the stirring alginate solution in a 1:2 molar ratio. Excess of 6-aminofluorescein was added to the mixture, and the reaction was allowed to proceed overnight at room temperature, protected from light. The fluorescein-labeled alginate was purified by dialyzing against a decreasing salt gradient in distilled water and lyophilized.
Degradation of oxidized alginate hydrogels
Alginate with theoretical degrees of oxidation from 0% to 5% was dissolved in Dulbecco's modified Eagle's medium (DMEM) to a concentration of 2%–10% (w/v). Alginate hydrogels were crosslinked by the addition of a sterile 1.22 M calcium sulfate slurry at 4% (v/v) of total gel. Gels were cast between two Teflon sheets separated by 2 mm for 45 min. Gels were then punched out using a 7-mm biopsy punch and washed with DMEM. The hydrogels were subsequently maintained at 37°C in DMEM. At designated time points, three gels from each condition were removed from media, frozen, and lyophilized. The dry mass of the gels over time was recorded to quantify the degradation rate of the alginate hydrogels.
Preparation of PEG-gelatin hydrogels containing oxidized alginate microfibers
Alginate microfibers were prepared by extruding 2% (w/v, unmodified alginate) or 3% (w/v, oxidized alginate) alginate solutions in DMEM through a 30-gauge Teflon needle into a stirring crosslinking bath composed of 1.0% (w/v) calcium chloride and 0.9% (w/v) sodium chloride. The fibers were removed from the crosslinking solution and equilibrated in a 20% (w/v) solution of PEG (MW=3 kDa) in DMEM at 37°C for 1 h. Preincubation in nonmethacrylated PEG prevents infiltration of PEG-DMA during fiber encapsulation, which will occlude pores following alginate degradation. The fibers were removed from the media and cut into approximately equal-size pieces using a scalpel. Fifteen microliters of 10%–20% (w/v) PEG-DMA and 3% (w/v) Gelatin-MA pregel solution in DMEM was added to a custom-made gel-casting mold capable of holding 50 μL of gel total. These samples were partially crosslinked by exposure to UV light for 2.5 min, in order to form a layer of gel at the bottom of the scaffolds to improve microfiber retention. An additional 20 μL of pregel solution was added, followed by the alginate microfibers. These samples were fully crosslinked by exposure to UV light for an additional 5 min. The scaffolds were then transferred to fresh DMEM and maintained at 37°C. Hydrogels containing unmodified alginate fibers, that is, not hydrolytically degradable, were used to determine whether fiber degradation was necessary for cell transfection. As a direct comparison, fiber degradation was achieved by the addition of 8 mM ethylenediaminetetraacetic acid (EDTA) to the cell culture media.
Gene transfection in PEG-gelatin hydrogel scaffolds
HEK 293 cells were expanded in monolayer culture in DMEM supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S) at 37°C and 5% CO2. DNA NPs were formed by mixing plasmid DNA solution with PBAE polymer solution (D32-122) in 25 mM sodium acetate at pH 5.5, using a polymer/DNA ratio of 10:1 (w/w). Two reporter DNA plasmids were used, including a Gaussia luciferase plasmid (New England BioLabs, Ipswich, MA) and a green fluorescent protein (GFP) plasmid (Clontech, Mountain View, CA). Prior to lyophilization, sucrose was added as a cryoprotectant to the NP solution to reach a final concentration of 15 mg/mL. HEK cells were trypsinized and encapsulated at a concentration of 5×106 cells/mL in NP-containing, 4% oxidized alginate hydrogel solution (3% w/v) to make alginate microfibers. The alginate microfibers containing cells and NPs were then encapsulated in bulk PEG-gelatin hydrogels. As a control group, HEK cells and DNA NPs were also directly encapsulated in bulk PEG-gelatin hydrogels without sacrificial porogens, by mixing cells and NPs with the pregel polymer solution at a concentration of 5×106 cells/mL and crosslinking by exposure to UV light for 5 min. All groups were maintained in DMEM plus 10% FBS and 1% P/S at 37°C and 5% CO2. After 24 h, the scaffolds were transferred to new 24-well plates to remove the influences of the cells that might have been released outside of the scaffold during microfiber degradation. To quantify transfection efficiency inside hydrogels over time, luciferase DNA plasmid was used, and the cell culture medium was collected and replaced on days 2, 4, and 6 postencapsulation. Transfection efficiency was measured by assaying the luciferase activity from the collected culture medium using a BioLux Gaussia Luciferase Assay Kit (New England BioLabs). The luciferase data for the scaffolds with microfiber conditions was normalized by cell number using an AQueous One Cell Titer Assay (Promega, Madison, WI) to account for differences in cell retention efficiency among different samples. To compare the transfection efficiency among different groups, the luciferase activity from porogen-containing hydrogels was normalized by the cell number within the scaffolds after microfiber degradation. To determine the cell retention efficiency after the microfiber porogen degradation, HEK cells were encapsulated in the microfiber-containing scaffolds, and incubated in culture medium for 24 h to allow microfiber degradation and cell release. The cell number within the scaffold versus the cell number that fell out to the bottom of the tissue culture plates was then quantified using a PicoGreen dsDNA Assay Kit (Invitrogen, Carlsbad, CA) following three freeze-thaw cycles at −80°C to disrupt the cell membrane.
Monitoring cell morphology within microfibers and hydrogels using fluorescence microscopy
To visualize cells encapsulated inside alginate microfibers, HEK cells were labeled using Hoechst 33342 dye and then encapsulated into fluorescein-labeled alginate fibers following the procedure described earlier. To visualize cell-containing microfibers within 3D scaffolds, the fluorescently labeled microfibers were encapsulated into PEG/gelatin hydrogels in which the gelatin was rhodamine labeled. To visualize cell release from microfibers, HEK cells were first encapsulated in 4% oxidized alginate microfibers, and then into rhodamine-labeled gelatin/PEG scaffolds as earlier. All samples were cultured for 6 days and then fixed in 4% paraformaldehyde for 15 min at 37°C. To stain the cytoskeleton using immunofluorescence, cells were permeabilized with 0.1% Triton-X for 30 min, followed by incubation with FITC-conjugated anti-α-tubulin (Invitrogen). Hoechst 33342 was used as a nuclear counterstain. Images were captured on a fluorescent microscope (Axio Observer Z1; Zeiss, Thornwood, NY).
Scanning electron microscopy
Microfibers and bulk hydrogels were also imaged by SEM. To compare the morphology change before and after porogen degradation, images were taken immediately after fabrication or following 2 days of incubation in serum-containing medium. Scaffolds were washed in ddH2O for 30 s prior to imaging. Imaging was performed using a variable-pressure SEM (Hitachi 3400N, Tokyo, Japan) equipped with a Peltier Coolstage (Deben, Woburn, MA).
Mechanical testing
To measure the stiffness of 3D bulk hydrogels, unconfined compression testing was performed on bulk hydrogels without microfibers containing varying PEG-DMA concentrations (10%, 15%, or 20%) (w/v) as described previously. 27 Gelatin-MA concentration was kept constant at 3% (w/v) to maintain a comparable level of biochemical cues within 3D hydrogels. Briefly, hydrogels without microfibers were formed in a cylindrical mold (6.5 mm in diameter and 1.5 mm in thickness), and placed in phosphate-buffered saline (PBS) for 24 h to reach swelling equilibrium prior to mechanical testing. Unconfined compression tests were conducted using an Instron 5944 materials testing system (Instron Corporation, Norwood, MA) fitted with a 10-N load cell (Interface, Inc., Scottsdale, AZ). All testing was performed in PBS solution at room temperature. A preload of ∼2 mN was applied before each test. The upper platen was then lowered at a rate of 1% strain/s to a maximum strain of 30%. Load and displacement data were recorded at 100 Hz. The compressive modulus was determined for strain ranges of 0%–10%, 10%–20%, and 20%–30% from linear curve fits of the stress versus strain curve in each strain range.
Statistical analysis
Minitab™ (Minitab, Inc., State College, PA) software was used for statistical analysis. One-way analysis of variance (ANOVA) with a Tukey's post hoc analysis was used to determine statistical significance between groups while a paired t-test was used to directly compare two groups. A value of p<0.05 was considered statistically significant.
Results
To determine the optimal formulation of alginate porogens with a desirable degradation rate, alginate with varying degrees of oxidation was prepared. Ionically crosslinked alginate hydrogels were produced from alginates with varying degrees of oxidation (0%, 3%, 4%, and 5%) and incubated in serum-containing cell culture medium for up to 100 h. The change in dry mass of the alginate hydrogels was recorded over time to monitor rate of degradation (Fig. 2). The mass of unoxidized alginate remained constant over time, whereas all groups based on oxidized alginate degraded fully within 50 h. Increasing the degree of oxidation of alginates from 3% to 4% and above dramatically accelerated the alginate degradation, with the 5% oxidized hydrogel scaffolds fully degrading within 2–3 h. A desirable porogen should degrade fast enough to minimize degradation of the encapsulated polymeric NPs, but still give cells sufficient time to adhere to the internal surface of microchannels after the porogen removal. The 4% oxidized alginate formulation meets these criteria, and was selected for fabricating hydrolytically degradable, microfiber-like porogens in further experiments.

Degradation profile of hydrolytically degradable alginate with varying degrees of oxidization (3%, 4%, and 5%), as quantified by percentage of weight loss. Unmodified alginate was included as a control. Inset: Degradation profile within the first 8 h showed rapid degradation of alginate oxidized at 4% and 5%.
To fabricate alginate solutions into microfiber-like porogens, the alginate solution was extruded into a calcium chloride bath under constant stirring. The as-spun microfibers demonstrated an average diameter of ∼250 μm, as shown by SEM characterization (Fig. 3A) and light microscopy (Supplementary Fig. S1; Supplementary Data are available online at
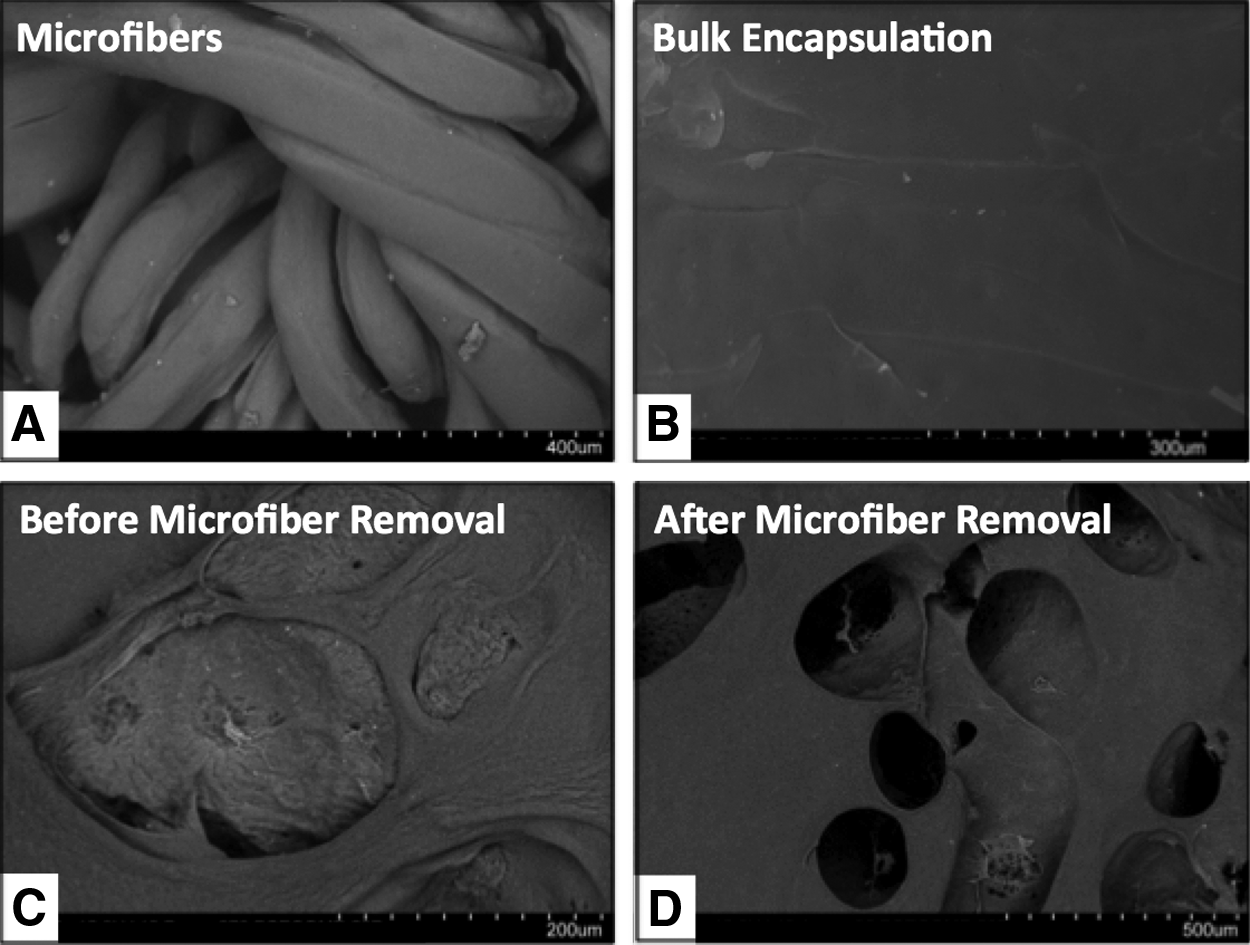
Scanning electron microscopy images of microfiber porogens and bulk hydrogel scaffolds.
We then assessed the effects of hydrolysis-triggered porogen degradation on the viability and morphology of the encapsulated cells. HEK cells encapsulated within the alginate microfibers demonstrated high cell viability following encapsulation, as shown by live/dead staining (Supplementary Fig. S2). Cells were uniformly distributed within fluorescein-labeled alginate microfibers (Fig. 4A). Immediately following incorporation into PEG-DMA/Gelatin-MA hydrogels, cells remained encapsulated within intact alginate microfibers (Fig. 4B). After 6 days of culture, substantial void spaces formed within the bulk hydrogels, with HEK cells released from alginate microfibers, adhered, and spread within the voids left by the degraded microfibers (Fig. 4C).
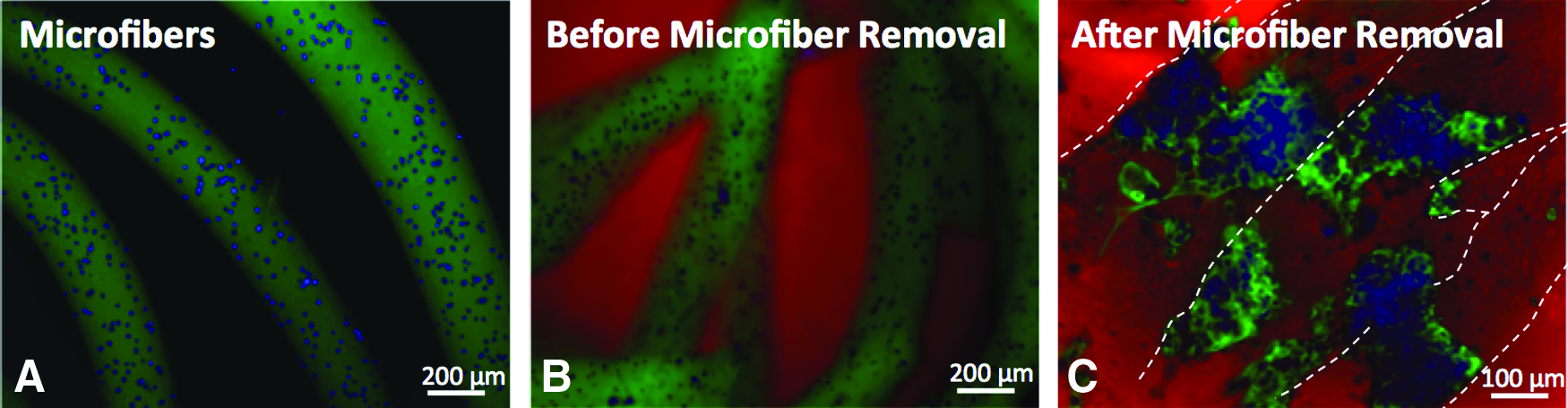
Fluorescence images of cells in sacrificial microfibers encapsulated within bulk hydrogels.
To examine the effects of sacrificial porogen incorporation on nonviral gene delivery inside hydrogels with a broad range of stiffness, alginate microfibers containing HEK cells were encapsulated in PEG-DMA/Gelatin-MA hydrogels with stiffness varying from 9 to 197 kPa. To quantify the transfection efficiency in bulk hydrogels or porogen-containing hydrogels, a bioluminescence assay was utilized to measure the amount of secreted Gaussia luciferase (New England BioLabs) in the culture medium, and bioluminescence intensity was normalized by cell number. The percentage of cells retained within the scaffolds after fiber degradation was comparable across hydrogels with different stiffness (Supplementary Fig. S3). Our results also showed that transfection efficiency is highly dependent upon the close proximity between the NPs and cells, as well as the distributions of cells and NPs within the hydrogels. Significant transfection was observed only when the NPs were co-encapsulated with the HEK cells inside the alginate fibers. In contrast, no transfection was observed when NPs were encapsulated within the bulk hydrogels or added in the medium, when the cells were encapsulated in the degradable alginate microfiber porogens. Further, cells must be released from the fibers to achieve efficient transfection (Supplementary Fig. S4). Cells encapsulated in microfiber porogens generally resulted in significantly higher transfection efficiency than cells encapsulated directly within the bulk hydrogels (Fig. 5). While the amount of transfection decreased with increasing hydrogel stiffness when cells were encapsulated within bulk hydrogels, the opposite trend was observed when cells were released from the sacrificial porogens. For example, at the highest hydrogel stiffness (197 kPa), cells encapsulated in porogens resulted in ∼15-fold higher luciferase activity compared with the cells encapsulated in bulk hydrogels.
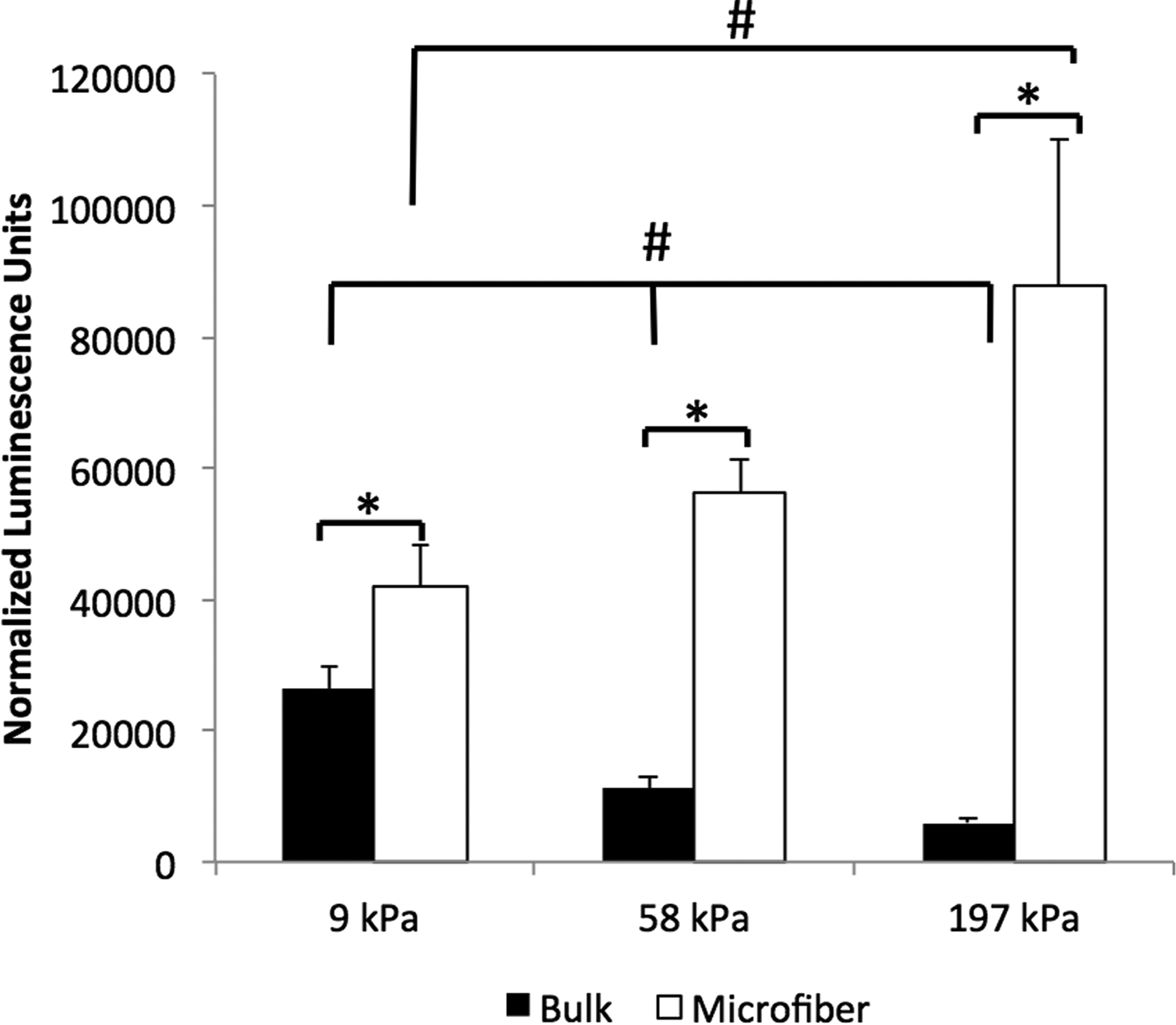
Transfection efficiency of the HEK cells encapsulated directly within bulk hydrogels or in sacrificial microfibers inside hydrogels with varying stiffness (9, 58, and 197 kPa). Transfection efficiency was reported using luminescence intensity of secreted Gaussia luciferase normalized by cell number. Data represent cumulative luminescence over 6 days. Luminescence values for untransfected cells were below 1000. Error bars represent±standard error of the mean. *,#p<0.05.
Discussion
When cells are encapsulated in hydrogels, the physical constraint of the polymeric network may limit cell proliferation. Incorporating cell adhesive ligands or balancing cell migration with matrix degradation has been shown to increase gene delivery efficiency inside hydrogels, likely by increasing cell proliferation.12,14 Incorporation of sacrificial porogens into hydrogels allows formation of void space within the gels, which may facilitate cell proliferation and migration, which are particularly important for polymeric NP-based nonviral gene delivery. 28 When the cells are encapsulated inside 3D hydrogels, increasing hydrogel stiffness typically correlates with decreased mesh size, which results in decreased cell proliferation and nonviral gene delivery efficiency. 12 By releasing cells from sacrificial porogens within hydrogels, we removed the physical constraint of the bulk hydrogel network on cells, and released cells sense hydrogels more as a 2D substrate. Further, microchannel formation will result in improved nutrient accessibility, which may increase cell proliferation and transfection efficiency. Indeed, our results showed increased gene delivery efficiency using sacrificial porogens as the hydrogel stiffness increased, which is similar to that observed when cells are cultured on 2D substrates. 29 Such increased gene delivery efficiency was correlated with increased cell proliferation on stiffer substrates, and the effect of matrix stiffness on nonviral gene delivery may differ among cell types due to varying sensitivities to matrix rigidity. 20 Compared with cells encapsulated inside bulk hydrogels, we observed up to a 15-fold increase in transfection efficiency in stiff hydrogels (197 kPa) using sacrificial porogens. Mechanical stiffness has been shown to regulate stem cell differentiation. 1 Mesenchymal stem cells cultured on hydrogels with tissue-mimicking stiffness demonstrated higher tendency to differentiate toward specific tissue lineages. The hydrogel stiffness chosen in the current study is within the range previously used to study musculoskeletal differentiation of stem cells. 2 The lower range (9 kPa) mimics muscle stiffness, while the intermediate and upper values (58 and 197 kPa) mimic bone precursor osteoid at various stages of development.1,30 In our porogen system, cells released into the newly formed void space can sense the internal surface of the macropores, and respond to the hydrogel stiffness more as if they were cultured on 2D substrates. Together, these results demonstrate that our platform allows efficient nonviral gene delivery inside hydrogels across a broad range of stiffness, which would be useful for promoting lineage-specific stem cell differentiation by employing both mechanotransduction and nonviral gene delivery.
Choosing a cell-friendly stimulus for porogen removal is an important aspect to consider for cell-containing scaffolds. We have chosen hydrolytically degradable alginate to serve as sacrificial porogens, as alginate can be easily processed into microspheres or microfibers, while supporting direct cell encapsulation with high cell viability. While alginate porogens can be removed using EDTA as a chelator to remove diavalent cations, EDTA may interfere with integrin-mediated cell adhesion and cause potential cytotoxicity. Therefore, we have chosen to use hydrolytically degradable alginate as our porogen material, which was prepared by oxidizing alginate with sodium periodate. This reaction results in the cleavage of the diol linkages within the uronic acid monomers of the alginate, promoting an open-chain conformation that is susceptible to hydrolysis. 21 Using the alginate with an optimized degradation rate, HEK cells and NPs can be co-released and attached within macropores inside bulk hydrogels over the course of 24 h. In the present study, PEG was chosen as the bulk hydrogel material given its tunable biochemical and mechanical properties, as well as its broad use in tissue engineering applications. Given that porogen removal is independent from the properties of the bulk hydrogels, this strategy of co-releasing cells and NPs from microfiber porogens could have general applicability to enhance nonviral gene delivery inside 3D hydrogels, irrespective of the type of hydrogel chosen for the bulk scaffolds.
A recent study has investigated the inclusion of spherical macropores into hydrogels to facilitate cell ingrowth and enhance viral-based gene delivery. Temperature-sensitive gelatin microspheres were used as porogens to enhance cell infiltration and subsequent lentiviral-mediated gene delivery in vivo. 17 The authors reported that prolonged gene expression only occurred when cells and lentivirus were co-encapsulated inside the bulk hydrogels, but not when cells were encapsulated inside bulk hydrogels while lentivirus was released from porogens. These results suggest that spatial distribution of cells and transfection reagents may also influence the gene delivery efficiency inside hydrogels. We examined the effects of varying the location of cells and polymeric NPs and found that close proximity of the two within sacrificial microfibers was required to achieve efficient gene delivery (Supplementary Fig. S4). The previous study used viral vectors and spherically shaped porogens, and the porogens were only used to create macropores and release viral vectors without cell delivery. Our report here focused on nonviral gene delivery using biodegradable polymeric NPs, and this platform could be particularly useful for tissue engineering applications in which nonviral-based gene delivery is preferred. The biodegradable polymeric NPs used in this study are formed using a PBAE, a hydrolytically degradable polymer. Previous studies have demonstrated that PBAEs can achieve high transfection both in vitro and in vivo.24,31,32 Additionally, transfection efficiency can be further tuned by varying the chemical structures of PBAEs. 25 In the current study, we have chosen a PBAE backbone containing six consecutive carbons, which led to enhanced transfection within hydrogels in our previous work. 13
One unique aspect of this study is that we employ porogens not only for creating macropores within the hydrogels, but also for co-releasing cells and polymeric NPs within microchannel-like structures. Creating macropores within bulk hydrogels allows enhanced cell infiltration, nutrient diffusion, and blood vessel penetration. By using porogens for releasing cells and biological signals, we can also achieve spatial patterning of cells and biological signals. Previous work on introducing porogens within bulk scaffolds generally employs spherically shaped porogens.16,22,33,34 Unlike the previous strategies, here we have chosen microfiber-like porogens to deploy cells within interconnected microchannel-like structures. This could aid in engineering tubular-like tissue structures such as blood vessel networks. Further, using microfibers allows better interconnectivity among the macropores without the need of increasing porogen concentration. In contrast, achieving interconnectivity among the macropores for microsphere-like porogens would require the use of high density of microspheres. This would substantially compromise the overall stability and mechanical strength of the scaffold, which is undesirable for engineering tissue types in which mechanical strength is important.
Conclusions
Here we report a method for enhancing nonviral gene delivery efficiency inside 3D hydrogels using sacrificial microfibers to create macropores and as delivery vehicles for co-releasing cells and polymeric NPs. Using the optimized formulation of alginate microfibers, we showed effective degradation of the microfibers within the bulk hydrogels, which support high cell viability and adhesion upon release. We further demonstrated that close proximity between cells and NPs within the porogen is required for efficient gene delivery. Compared with cells encapsulated directly within bulk hydrogels, sacrificial microfiber-mediated delivery resulted in an increase in transfection efficiency of up to 15-fold in hydrogels across a broad range of stiffness (9–197 kPa). Opposite to the trend observed in bulk hydrogels, increasing hydrogel stiffness led to enhanced gene delivery efficiency using sacrificial microfiber-mediated delivery. This platform reported herein may be applicable for promoting desirable cell fate and tissue regeneration by allowing efficient nonviral gene delivery inside hydrogels within a broad range of stiffness.
Footnotes
Acknowledgments
The authors would like to thank the Stanford Bio-X Interdisciplinary Initiative grant and the Basil O'Connor Starter Scholar Research Award from March of Dimes Foundation for funding.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.