
Abstract
Despite the development of a myriad of anticancer drugs that appeared promising in preclinical ovarian cancer animal models, they failed to predict efficacy in clinical testing. To improve the accuracy of preclinical testing of efficacy and toxicity, including pharmacokinetic and pharmacodynamic evaluations, a novel animal model was developed and characterized. In this study, murine ID8 (epithelial ovarian cancer [EOC]) cells as injected cell suspensions (ICS) and as intact cultured monolayer cell sheets (CS) were injected or surgically grafted, respectively, into the left ovarian bursa of 6–8 week-old, female C57BL/6 black mice and evaluated at 8 and 12 weeks after engraftment. Tumor volumes at 8 weeks were as follows: 30.712±18.800 mm3 versus 55.837±10.711 mm3 for ICS and CS, respectively, p=0.0990 (n=5). At 12 weeks, tumor volumes were 128.129±44.018 mm3 versus 283.953±71.676 mm3 for ICS and CS, respectively, p=0.0112 (n=5). The ovarian weights at 8 and 12 weeks were 0.02138±0.01038 g versus 0.04954±0.00667 g for ICS and CS, respectively (8 weeks), p=0.00602 (n=5); and 0.10594±0.03043 g versus 0.39264±0.09271 g for ICS and CS, respectively (12 weeks), p=0.0008 (n=5). These results confirm a significant accelerated tumorigenesis in CS-derived tumors compared with ICS-derived tumors when measured by tumor volume/time and ovarian weight/time. Furthermore, the CS-derived tumors closely replicated the metastatic spread found in human EOC and histopathological identity with the primary tumor of origin.
Introduction
E
The most common EOC histological subtype, accounting for >50% of ovarian epithelial malignancies, is serous ovarian carcinoma. 3 Due to the lack of early detection tools, the vast majority of serous ovarian carcinomas (>80%) are diagnosed at a more advanced stage 4 (stages III–IV), where the 5-year survival rate remains only 9–34%. 5 Most of these (>50%) are classified as “high-grade” tumors based on their degree of nuclear atypia and high mitotic index. 6 High-grade serous ovarian carcinomas (HGSOC type II) are characterized from other subtypes both for their aggressive nature and because they may harbor unique genetic alterations, including TP53 and the DNA repair genes BRCA 1 and 2. In contrast, clear cell, endometrioid, low-grade serous, and mucinous ovarian carcinomas typically present as indolent low-grade neoplasms (type I tumors) with somatic mutations in genes such as KRAS, BRAF, ERBB2, PTEN, CTNNB1, and PIK3CA. 7
The ideal ovarian cancer animal model should closely replicate human disease etiology, genetic characteristics, tumor heterogeneity, and mimic interactions with the host immune system and relevant microenvironments. Various reviews8,9 described different techniques to establish animal cancer models. Early models used chemical carcinogens to induce tumorigenesis,10–12 modeling later advanced with the viral introduction of oncogenes inducing spontaneous tumors.13,14 Though these models may enable spontaneous tumorigenesis, genetically engineered cancer mouse models 15 are associated with extremely high cost and difficulty for scalable reproducibility. Xenograft models utilizing human cancer cell lines are predominantly used to establish tumors in immune-deficient (nude) mice, and they are frequently created by subcutaneous, nonorthotopic single-cell injections. Therefore, the tumor site lacks the appropriate microenvironment and immune system interactions deemed relevant for the development of human EOC. 16 Orthotopic tumor origination may reproduce a more characteristic, metastatic spread of disease as well as a therapeutic response. Alexander reviewed the problems and shortcomings of current animal cancer models, 17 pointing out that the architectural and cellular complexity of actual tumors, including innate vasculature, stromal and inflammatory components in a specific anatomic location, cannot be faithfully replicated in cell culture. Furthermore, the xenograft–tumor–host interactions in a subcutaneous microenvironment are quite different than those found in the actual tissue of origin, thereby impacting drug response and therefore treatment outcomes.
In this project, we exploit the feasibility of engineered cell sheet (CS) cultures 18 as a novel concept for orthotopic ovarian tumor tissue engraftment in mice. CS tissue engineering offers several advantages compared with the conventional injection of single-cell suspensions. Tumor cell delivery occurs in the form of an intact cell monolayer preserving all cell–cell junctions, receptors, and the relevant extracellular matrix (ECM). Commercial plastic cell culture dishes grafted with a thin layer of temperature-responsive poly(N-isopropylacrylamide) (PIPAAm) enable culture and recovery of intact CSs by simply reducing the temperature from 37°C to room temperature (20–25°C).19–21 This technique avoids harsh enzymatic treatments damaging cells during their dissociation into suspensions. Noncancerous CS cultures adhere rapidly to numerous different host tissues,22–25 and promote improved graft quality and survival compared with tissue derived from injected cell suspensions (ICS). 26 For example, transplanted hepatocyte CSs express significantly greater hepatocyte-specific proteins, and demonstrate increased drug metabolism compared with hepatocyte ICS. 22 In cardiac repair experiments using transplanted myoblast CS tissues, increased formation of neocapillaries with reduced fibrosis was seen when compared with tissues derived from myoblast ICS. 27 Additional reports also confirm that large central necrotic areas are present in ICS-derived tissue constructs, whereas CS constructs exhibit homogenous stratified tissues without areas of observable necrosis. 28 Standard operating procedures have been established for the processes involved in CS tissue engineering, which have already advanced to be used in a recent regenerative medicine clinical trial.29,30 Therefore, this technique now offers standardized, reproducible, operator-independent protocols that are translatable from noncancerous to cancerous cells and potentially will serve as preclinical animal testing platforms more efficiently and reliably screening novel anticancer therapeutics.
This study describes the development and characterization of an engineered EOC-CS animal tumor model. While CS techniques have been established for the in vitro and in vivo reconstruction of various other noncancerous tissue types,31–33 this research represents an original effort to design and construct a novel animal model of EOC using CS technology. Its utility will be evident by demonstrating adequate tumorigenesis, improved tumor–tissue integration, greater fidelity to human ovarian tumor physiology and histology, and functionality in an immune-competent animal.
Materials and Methods
Cell culture
Murine ovarian ID8 cancer cells, a kind gift from Dr. G. Coukos (University of Pennsylvania), were derived from C57BL/6 mouse ovarian surface epithelial cells 34 ; maintained in culture complete medium, Dulbecco's modified Eagle's medium (Sigma Aldrich) supplemented with 4% fetal bovine serum (Denville Scientific), 1% penicillin and streptomycin (Sigma Aldrich), and 1% insulin–transferrin–selenium (Invitrogen) at 37°C; and incubated in a humidified atmosphere of 5% CO2. When ID8 cell cultures reached subconfluency, they were treated with trypsin-EDTA and re-suspended in phosphate-buffered saline (PBS) for both ICS injection and CS fabrication.
Preparation of ID8 CSs
Temperature-responsive polymer (PIPAAm)-grafted cell culture dishes were provided (UpCell™; CellSeed) and used as previously described. 19 Through slight ambient temperature reduction, these dishes change surface properties and release attached cells without use of conventional dispase or trypsin protocols. This permits ease of culture and subsequent reliable collection of intact CSs. ID8 cells harvested using traditional enzymatic trypsin treatment on conventional cell culture plastics (Falcon) were transferred and seeded into 35 mm-diameter temperature-responsive culture dishes (CellSeed) (2×106 cells per one dish) in a humidified incubator with 5% CO2 at 37°C for 24 h. When cells reached confluency, culture dishes were placed inside a laminar flow hood at room temperature (20–23°C) for 5–10 min to enable cells to detach from the surface in the form of an intact CS layer. ID8 CSs for implantation were prepared with a diameter of 6 mm. ID8 CSs were sectioned in half using a scalpel (Grafco Feather Sterile Scalpels; Disposable #11). To ensure equivalency of cell numbers used in ICS and in CS, numbers of cells (1×106) were counted before injection/transplantation and reconfirmed for CS after trypsinization of a separate dish not used for transplantation. The CS was then siphoned using a 10 μL pipette for transfer from the culture dish to animal ovarian tumor sites that were surgically prepared as described next. The remaining part of ID8 CS was preserved in paraformaldehyde for additional histological analyses.
Animals
Six- to 8-week-old female C57BL/6 mice (Jackson Laboratories) were housed and treated under standard conditions in the Animal Facility of the Center for Comparative Medicine at the University of Utah according to approved protocols and guidelines of the Institutional Animal Care and Use Committee (IACUC).
Tumor initiation by ID8 ICS
ICS of ID8 cells in PBS were prepared immediately before an injection into animal tumor sites. Animals were randomly divided into nine groups with n=3–5 per group. All animals undergoing surgical procedures were weighed before anesthetic injections (intraperitoneal xylazine–ketamine, 0.1 mL/10 g body weight). Each animal was placed in prone position and a dorsal incision of ∼1.5 cm in length, slightly to the left of the midline, was made. The dermis was separated from underlying tissues, and a smaller incision was made through the dorsal fascia flat muscle to access the abdominal peritoneum. The left kidney was identified, and the area immediately below the kidney was dissected to locate the left ovary, which was then externalized through the incision. The control group (n=3) received only a sham surgical procedure (SSP) without any injections into the ovary, and a second control group (n=3) received an injection of 10 μL PBS (control) with SSP into the left ovarian bursa. All other groups (n=5) received an injection of 1×106 ID8 cells (10 μL cell suspension in PBS) into the left ovarian bursa. ICSs were directly injected into the ovarian bursa using a Hamilton syringe (30G needle). After cell inoculation, the ovary was placed back into its original position in the peritoneal cavity. The back wall and skin incisions were sutured separately using 4-0 sutures. Control groups were sacrificed at 14 weeks after the surgical procedure. ID8 ICS injected groups were sacrificed at 8 weeks (n=5), 12 weeks (n=5), and 14 weeks (n=5), respectively.
Tumor initiation by ID8 CS transplantation
Animals were randomized into four groups for sacrifice at 1 week (n=4; one animal was lost during anesthesia), 4 weeks (n=5), 8 weeks (n=5), and 12 weeks (n=5). To receive ID8 CS transplantations, animals were identically anesthetized and then, identical surgical site procedures were followed as described for animals in the ID8 ICS groups. Ovarian stromal tissue was accessed with a 1–1.5 mm incision to place ID8 CS. One half of a CS containing 1×106 cells was transplanted into the mouse ovaries using a 10 μL pipette. After allowing the transplanted CS to attach to the host ovarian tissue for about 2–3 min, the bursal incision was sutured closed using 4-0 sutures (Fig. 1A).
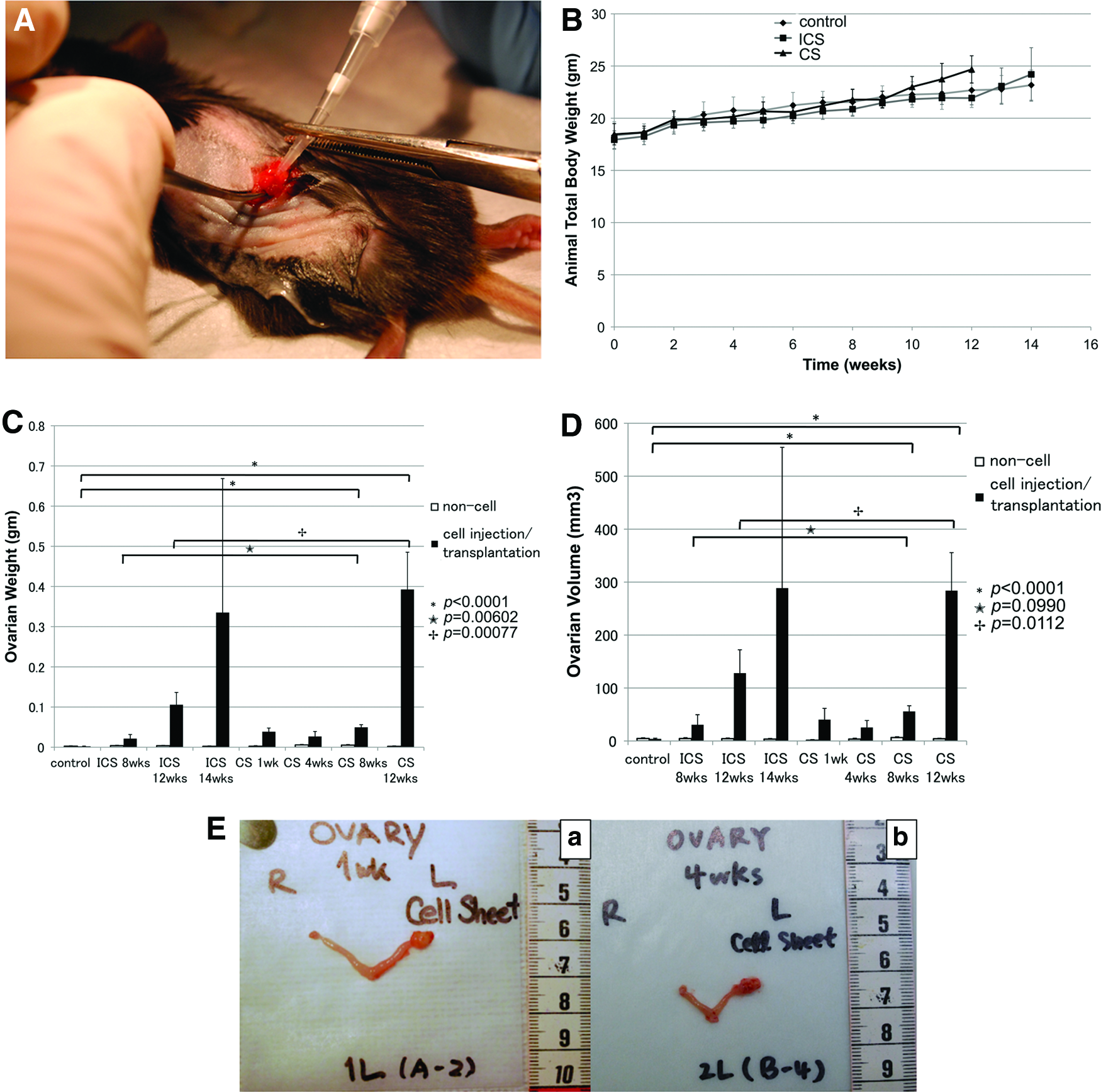
Ovarian volume and weight
All animals were sacrificed in accordance with the University of Utah IACUC-approved protocols. Oophorectomies were performed immediately after sacrificing mice at predetermined time points. Volumes and weights of the ovaries were captured by digital scale and caliper. Removed ovaries were formalin fixed, paraffin embedded and histopathological procedures were performed on a representative section of both tumor and normal ovary at each time point (at ARUP Laboratories).
Location and number of metastases
The location and number of metastases was determined. Every metastatic lesion in each affected organ was added to reach a final count for each organ and surface area (i.e., peritoneal sidewall) by visual observation.
Histopathology
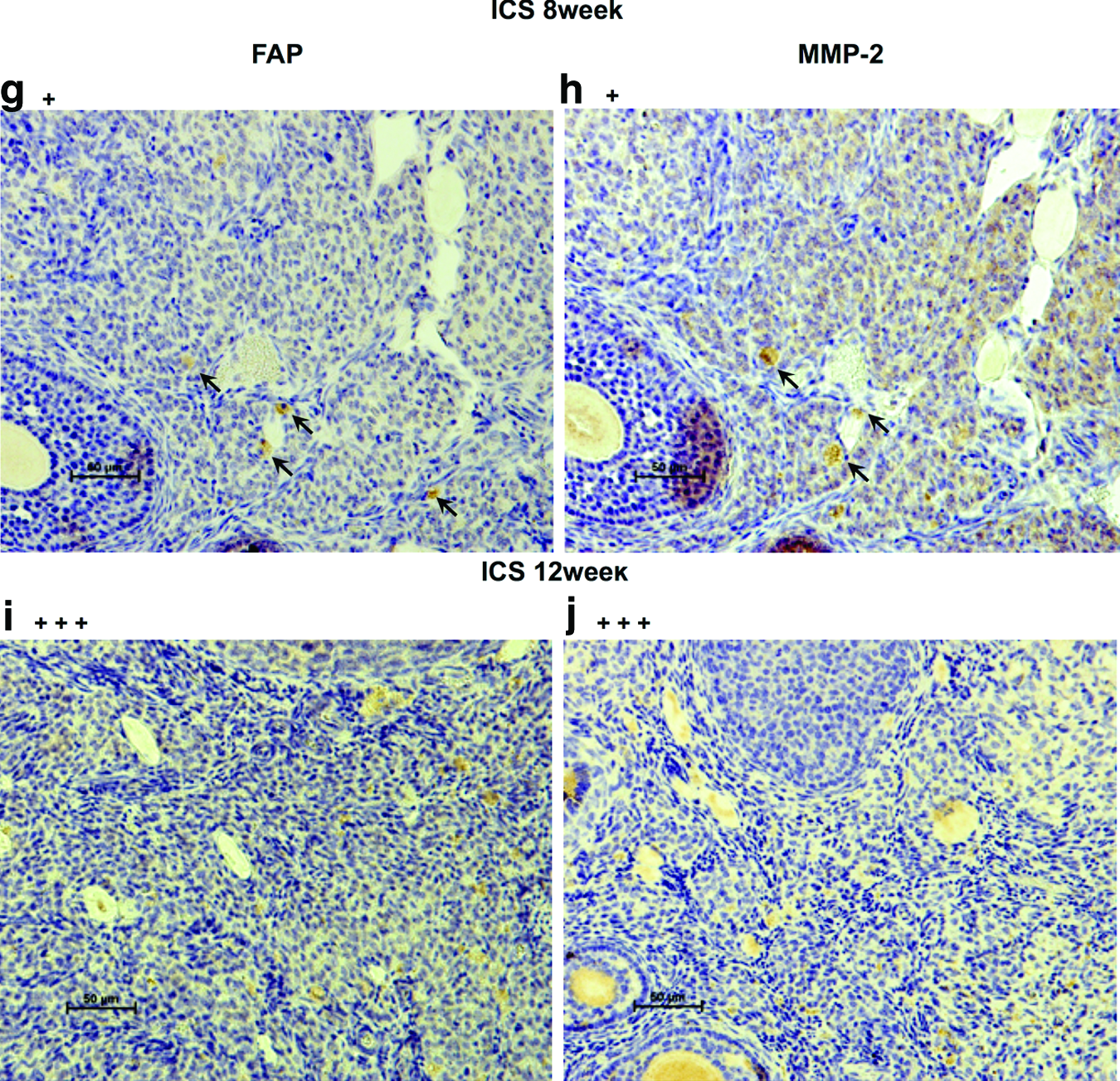
Hematoxylin–eosin (H&E) staining and immunostaining with antibodies for CD31, vascular endothelial growth factor (VEGF), fibroblast activation protein (FAP) (ab28244; Abcam), and matrix metalloproteinase-2 (MMP-2) (ab15102; Abcam) were performed on the ID8 CS before transplantation as well as on resected ICS- and CS-generated tumor tissues at each time point. Human FAP/seprase is a surface glycoprotein expressed on cancer-associated fibroblasts (CAFs) in the majority of epithelial cancers, including EOC. Since FAP overexpression in human tumor cells has been shown to promote tumor growth in animal models and relate to the function of MMP-2, we used anti-FAP antibody and anti-MMP-2 antibody to stain the resected tissues (CS-derived tumors at 1, 8, and 12 weeks and ICS-derived tumors at 8 and 12 weeks).
After a 24 h culture period, ID8 CS and resected tissues were fixed in 4% paraformaldehyde (Wako Pure Chemical) and routinely processed into paraffin-embedded sections. H&E staining was performed per conventional protocols. Formalin-fixed, paraffin-embedded tissue sections that were 4 μm in thickness were placed on positively charged slides and dried in air. After drying, slides were melted in a 60°C oven for 30 min before they were placed on an automated stainer (Ventana Medical Systems). Slides were deparaffinized with EZ Prep solution (Ventana Medical Systems) and pretreated with CC1 (cell conditioning 1) for 30 min (Ventana Medical Systems).
H&E staining was performed as per conventional protocols. Immunohistochemical staining for CD31 was performed on representative tumor sections at each time point, whereas VEGF staining was only performed on CS-derived tumor tissue at the 1-week time point (ARUP Laboratories). 35 Briefly, to evaluate microvessel density in tumor tissues, staining for CD31 and VEGF was performed. Each primary antibody (CD31/1:50, ab 28364, or VEGF/1:50, ab 28364; Abcam) was applied to the slides for 2 h at 37°C after treatment with secondary antibody (rabbit IgG; Sigma Aldrich). The detection of slides was performed on the Ventana XT automated system using the IView DAB detection kit, which is a streptavidin-HRP system, utilizing 3-3′ diaminobenzidine (DAB) as the chromogen. Slide counterstaining was performed with hematoxylin. Digital images were acquired through a digital camera that was equipped with a microscope (Olympus BH-2; Olympus America, Inc.). The percentage of CD31 staining area was analyzed using ImageJ 1.44o software (NIH), and a minimum of five high-power (400×) fields of view were evaluated for each tissue.
In staining for FAP and MMP-2, primary antibodies were 1/1000 and 1/200 dilution, respectively. Tissues were incubated overnight at 4°C followed by secondary antibody for 1 h at room temperature. Enzymatic staining for FAP and MMP-2 with the use of LSAB2 kit (Dako Cytomation) was applied according to the manufacturer-suggested protocol. The counterstaining of slides was performed with Hematoxylin. The intensity of positive immunostaining was graded as +++, ++, and + for strong, moderate, and weak results, respectively.
Statistical analysis
Values are expressed as mean±standard deviation. Data were plotted using Excel, and statistical analyses were performed using ANOVA and Student's t-test analysis (using JMP® software version9).
Results
Comparison of ID8 ICS-derived tumors versus ID8 CS-derived tumors
Total body weight (TBW) was measured twice to thrice per week starting on day 1 postsurgery (Fig. 1B). TBW increases were similar across all groups for the first 10 weeks. Starting at 10 weeks, TBW for the CS transplantation group (23±1 g) was slightly greater than for the ICS injection group (21.82±0.53 g). After reaching 12 weeks, a significant difference could be observed (24.68±1.304 g versus 21.93±0.65 g, ICS and CS, respectively; p<0.001, n=5). No significant difference was found between the ICS injection group and the CS transplantation group with regard to volume and weight of control ovaries (right side) not receiving any ID8 tumor cells (Fig. 1C, D).
Volume and weight of cancerous ovaries (left side) having received either ICS injections or CS transplantations significantly increased compared with contralateral, healthy ovaries (Fig. 1E). Ovarian weights of the CS-grafted groups were much higher than those of the control groups. The most significant increase in ovarian weight was noted after 8 weeks for the CS-transplanted groups (0.04954±0.00667 g) when ovarian weights increased more than twofold compared with tumors within the ICS groups (0.02138±0.01038 g) (p=0.00602). Further, a significant difference was found when comparing the ovarian weights of ICS-injected groups (0.10594±0.03043 g) with CS-transplanted groups (0.39264±0.09271 g) after 12 weeks (p=0.00077) (Fig. 1C). Furthermore, ovarian volume indirectly correlated to ovarian weight at the 8 weeks: 30.712±18.800 mm3 for ICS versus 55.837±10.711 mm3 for CS, p=0.0990, and 12 week; 128.129±44.018 mm3 for ICS compared with 283.953±71.676 mm3 for CS, p=0.0112) (Fig. 1D).
Metastases
On comparing the total numbers of metastases at the 8-week time point, animals with CS-derived tumors (n=42) had developed twice as many metastases as animals with ICS-derived tumors (n=19). The total metastatic count was only 150 for the five animals within the 12-week ICS group (n=161) and >400 among all five animals within the 12-week CS group (n=405) (Fig. 2A). On comparing the locations of metastases at the 8-week time point for animals in the ICS injection groups, metastatic dissemination was localized to visceral organs in close proximity of the ovary (i.e., kidney, fallopian with uterine tube, and spleen). After 12 weeks, lesions had spread further across the peritoneal cavity, including the liver and intestines. In contrast, CS-transplanted groups exhibited metastases on the peritoneal sidewall, liver, kidney, and diaphragm by 8 weeks. In addition, by 12 weeks, ascites had also developed in these animals and metastases had spread to the fallopian with uterine tube, spleen, intestine, and omentum (Fig. 2B). In each case, the foci of the extraovarian tumor identified were morphologically identical to the primary ovarian tumor. Similar to the ovarian tumor, the extraovarian tumors were characterized by high-grade malignant cells with minimal cytoplasm and atypical nuclei, which were arranged in variably sized nests and poorly formed glands (Fig. 2C).

Morphological examination for ID8 CSs in vitro
Each engineered CS included 2×106 ID8 seeded cells per one temperature-responsive culture dish of 35 mm diameter size. Cell populations displayed no significant morphological changes within 24 h before seeding in culture. A histological examination of ID8 CS demonstrated that cells showed irregular shapes and sizes, with scant cytoplasm, numerous mitotic figures, and nuclear atypia indicative of a typical malignancy (Fig. 3A).
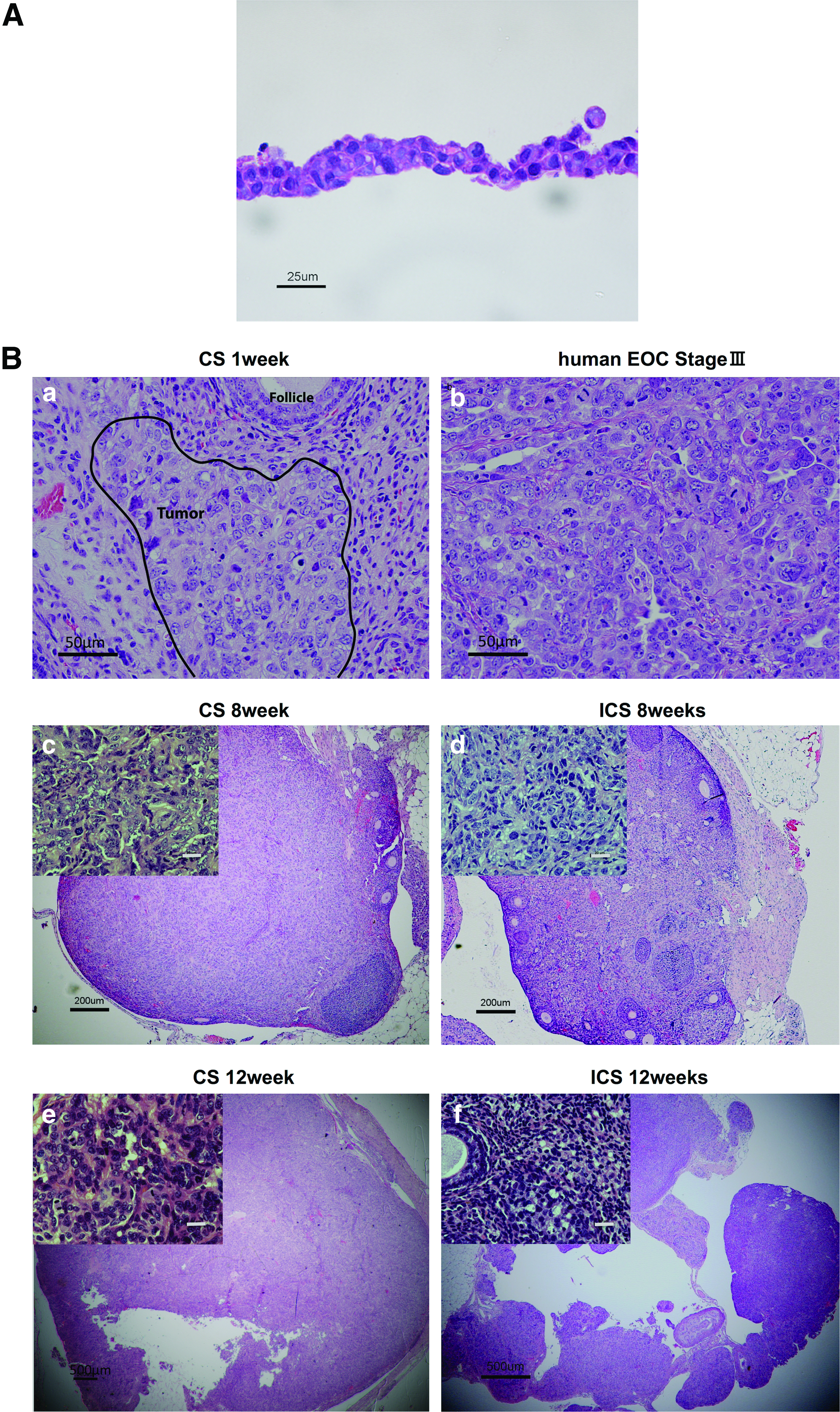
Morphological comparison of in vivo ID8 ICS-derived versus ID8 CS-derived tumors
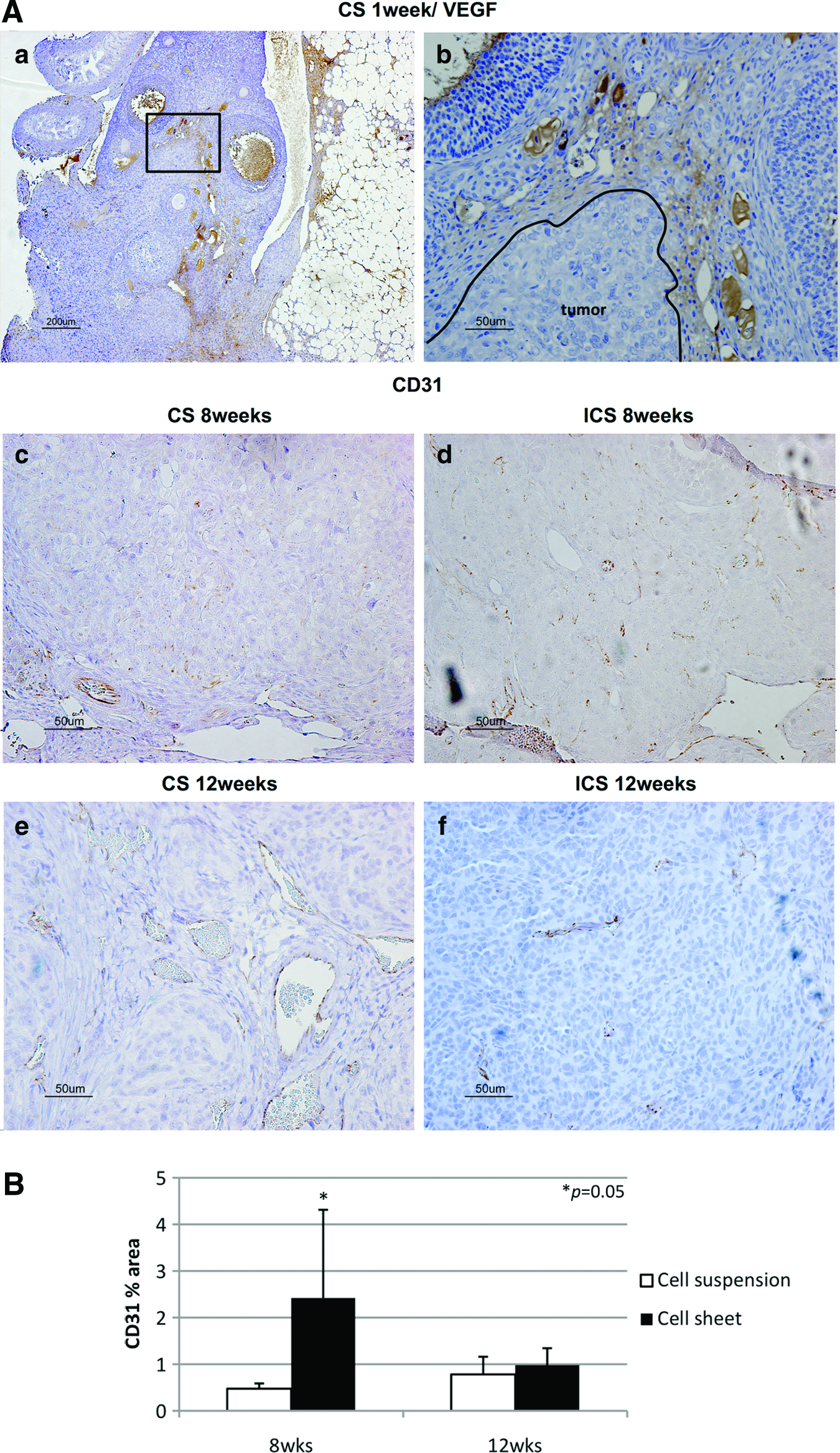
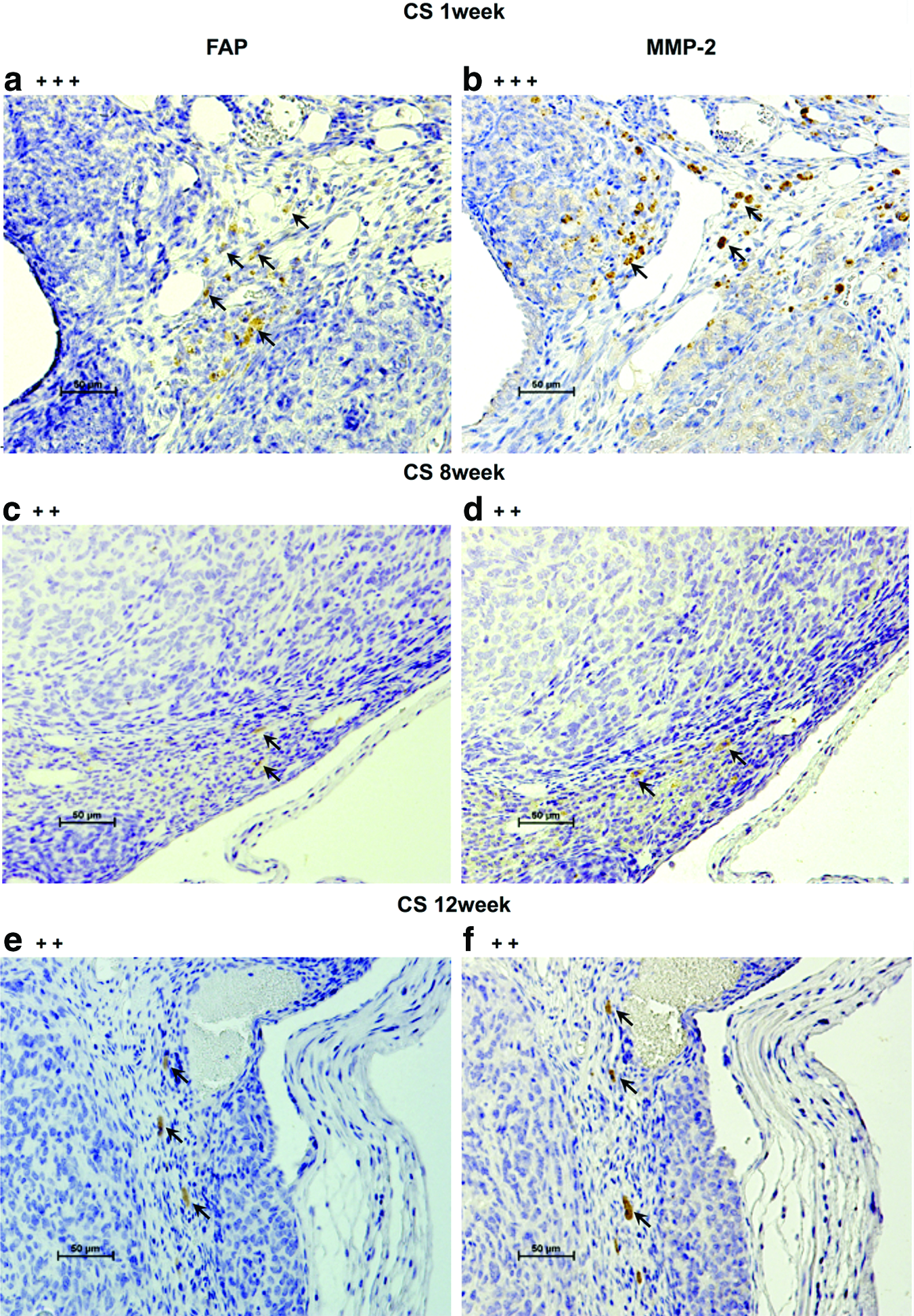
H&E staining confirmed ID8 cell-derived tumors in the ovarian bursa invading the ovary as early as 1 week after orthotopic CS transplantation. Transitional zones to normal, noncancerous tissues were clearly identifiable within the ovary (Fig. 3B-a) after 1 week with histological characteristics being similar to human spontaneous EOC (Fig. 3B-b). Furthermore, normal stromal tissue structure and histology in tumor were maintained and the ID8 cells displayed their typical characteristic features, including irregular shape and size of the cells, nuclear atypia, abnormal nuclear-cytoplasmatic ratios, and the presence of mitotic figures (Fig. 3B-a). Moreover, nearly all of the original normal structure of the ovary was replaced by metastatic tumor features in the CS-derived tumors when compared with the ICS-induced tumors at 8 weeks (Fig. 3B-c, d). After 12 weeks, the entire ovary was replaced by a tumor with malignant histology, and the volumes of CS-initiated tumors were rapidly growing compared with the tumors formed in ICS-treated ovaries (Fig. 3B-e, f). Further, CS-initiated tumors grew invasively, infiltrating the ovarian parenchyma. VEGF immunostaining of CS-derived tumors harvested at 1 week post-transplantation was positive (Fig. 4A-a, b). Immunohistochemical expression of the angiogenetic factor, CD31 revealed that the area including microvasculature at 8 weeks was stained to 0.475%±0.113% in ICS-derived tumors versus 2.420%±1.893% area in CS-derived tumors (p=0.05) (Fig. 4A-c, d, B). At 12 weeks, 0.782%±0.378% area versus 0.979%±0.364% area displayed positive CD31 staining in ICS-derived tumors and CS-derived tumors, respectively (p=0.43) (Fig. 4A-e, f, B). Tissue staining for FAP showed overlapping expression with MMP-2 in stromal cells of both the ICS-derived and CS-derived tumors (Fig. 5). CS tumors stained positive for FAP and MMP-2 strongest (+++) within the stromal tissue at 1 week post-CS transplantation (Fig. 5a, b), then gradually decreased over time (8, 12 weeks; ++) (Fig. 5c–f). FAP and MMP-2 expression increased from 8 weeks (+) to 12 weeks (+++) in the ICS-derived tumor (Fig. 5g–j).
Fibroblast activation protein (FAP) and matrix metalloproteinase-2 (MMP-2) expression. Scale bar is 50 μm (×200).
Discussion
In a previous study, we reported that ID8 tumorigenesis required ∼60 days post-ICS injection. 35 A sentinel finding in this study was a significantly shortened tumorigenesis requiring only 7 days post-transplantation when utilizing the CS approach in the same immune-competent animal model. This phenomenon may be due to the fact that CS technology retains cell–cell junctional complexes, expression of key membrane proteins and antigens, and the continued secretion of cytokines at the time of transplantation. Yamato et al. showed that E-cadherin and laminin-5 production, products of ECM, are maintained in the keratinocyte sheets harvested from temperature-responsive culture dishes. 36 The remarkably rapid ID8 CS fabrication within 24 h and significant reduction in time for in vivo tumor initiation, progression, invasion, and metastasis suggest that ECM deposition and expression of critical cancer inducing factors may be a key advantage provided by CS technology.
Generally, solid tumor consists of various other components besides cancer cells such as specific stroma, which is constructed from ECM, fibroblasts, inflammatory cells, and tumor-associated endothelial cells, cooperating to promote cancer cell propagation, survival, migration, invasion, and metastasis at tumor sites. 37 Due to the very limited space while manipulating CS with a 10 μL pipette, CSs were folded in some cases instead of being spread evenly as one single, flat layer onto the ovarian surface beneath the bursa. This modification led to inherent cell–cell interactions, including associated ECM, which were capable, despite folding, of maximizing cell retention and adherence within the bursal confines. Thus, the properties enhancing engraftment did not appear to be dependent on a single cell layer of CS adherence and were present despite marginal folding. This suggests that CS can be utilized in a variety of applications as tissue packets and not necessarily as a monolayer sheet.
The differentiation of ovarian carcinomas is very complex. 38 Tumors form polarized epithelia, papillae, cysts, and glandular structures. Morphological examination of ID8-CS in vitro displayed heterogeneity of various cell types, including both cancerous cells and fibroblast-like cells, very much akin to the findings in human EOC. Our in vivo experiments also showed similar morphological characteristics, including papillary tumor growth, multilayered epithelia, including stromal and capsular invasion (Fig. 6).
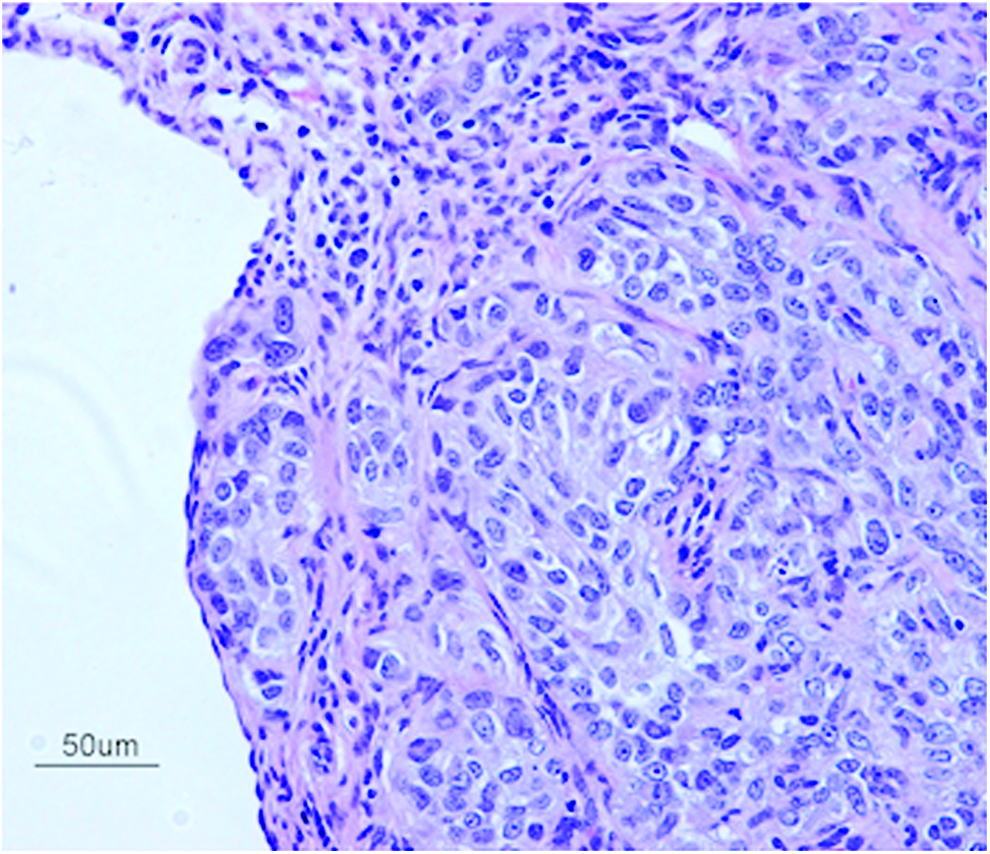
H&E staining reveals malignant epithelioid cells arranged in variably sized nests within CS-derived tumor at 8 weeks post-transplantation. The cells themselves have a minimal to moderate amount of eosinophilic cytoplasm and atypical nuclei exhibiting anisonucleosis, coarse chromatin, and variably conspicuous nucleoli. Scattered mitotic figures are noted. Scale bar is 50 μm (×200). Color images available online at
Noteworthy was the metastatic pattern in animals bearing orthotopically CS-transplanted tumors, which closely mimicked the metastatic spread in spontaneous human ovarian cancers. In general, serous ovarian cancer cells through surface shedding are disseminated within the peritoneal cavity carried by the peritoneal fluid to the peritoneum, diaphragm, and omentum.39,40 Ipsilateral kidney metastases were noted after 8 weeks in animals bearing CS-derived tumors, This type of metastatic spread is indicative for local surface shedding. After 12 weeks, animals, in addition to forming multiple metastases in various locations, also developed ascites, which was associated with hepatic and omental metastases (responsible for the animals' body weight gain starting around 10 weeks).
According to numerous reports, CAFs appear to enhance cancer cell invasion and metastasis. CAFs promote the ECM deposition, resulting in cancer fibrosis, and also MMP secretion, which is effective for resolving ECM, plays an important role in tumor growth and invasion. 41 Multiple other studies used anti-FAP antibody as a useful marker to detect CAFs42–44 in induced animal tumor models. Enhanced tumorigenesis of the CS-derived ovarian tumors may have been, in part, promoted by the natural orthotopic microenvironment, which provided FAP-expressing fibroblasts that potentially were induced by CS graft ECM factors.
MMP-2 immunostaining demonstrated a significant expression within the stroma of CS-derived tumors during the first week of tumorigenesis, compared with ICS-induced tumors. Torng et al. reported that stromal MMP-2, indeed, occurs early and may play a role in early EOC invasion having a key role in matrix remodeling by fibroblasts. 45 Furthermore, an association between MMP-2 and the invasion and metastasis of ovarian cancer has already been reported. 46 It is known that both force-mediated and protease-mediated matrix remodeling play a role in tumor invasion, and it has been reported that fibroblasts actively generate tracks through the neighboring stroma of tumor cells. 41 Our findings confirmed the expression of FAP and MMP-2 not only within the stroma surrounding the tumor but also in tissues not immediately adjacent to the tumor. This suggests that stromal fibroblasts may release MMP-2 and may also play a role in invasion and stromal remodeling, which is critical to initiate tumor invasion and cell migration. It appears that the ID8 CS-derived ovarian cancer model described here replicates the MMP-2-associated pattern of tumor invasion in the ovary.
Mesothelial cells covering organs located within the peritoneal cavity are purported to be the first line of defense to inhibit ovarian cancer cell adhesion and invasion.47,48 CS-derived, orthotopic, immunocompetent models of EOC could potentially offer a feasible new model that more closely replicates the human scenario. Mesothelial cells stimulate the migration of white blood cells in response to inflammatory mediators. After coculturing primary human mesothelial and ovarian cancer cells, both of which are extracted from ascites on ECM-coated culture dishes, increased attachment to ECM was observed for ovarian cancer cells compared with mesothelial cells. 48 In the case of using ovarian cancer spheroids, it has been shown that these use myosin-generated force when breaching through the mesothelial cell monolayer; moreover, Iwanicki et al. reported that ovarian cancer spheroids use myosin-generated force to breach and remove the mesothelial cell monolayer. 49 Further, the animal model described here benefits from the use of an immunocompetent host who offers an environment conducive to mesothelial cells, successfully stimulating the migration of white blood cells in response to secreted inflammatory mediators and possibly leading to the increased speed in orthotopic CS-derived tumor development compared with ICS-derived ovarian tumors. In CS-derived tumor-bearing animals, we also observed metastatic lesions throughout the peritoneal cavity, including the diaphragm at only 8 weeks post-transplantation. CS-derived ovarian tumors were breaking through the ovarian capsule at already 4 weeks post-transplantation, possibly facilitating the intraperitoneal tumor spread described earlier (Fig. 6).
Angiogenesis, indirectly documented by immunostaining for VEGF and CD31, was increased in CS compared with ICS tumors. VEGF expression was notably elevated in CS-derived tumors already at 1 week after engraftment. We believe that such VEGF expression in early stages of ovarian tumorigenesis may be an avenue for targeted therapy.50–52 As reported by Shimizu et al., 28 neovascularization was also detected in cardiac CS grafts at 3 weeks post-transplantation in vivo. Sekiya et al. reported that pulsatile cardiac CSs contained significant amounts of endothelial cells, and these endothelial cells formed a network-like construct within only 4 days in culture, 53 and demonstrated continuous VEGF expression in these cultured CS in vitro. These examples may, in part, explain the difference observed in ovarian volumes and weights in CS-derived compared woth ICS-derived tumors. Microvascular-like formations from the CS to host ovary may have been generated within the first 7 days after transplantation, resulting in improved tumor initiation, growth, invasion, and metastatic potential. In support of this hypothesis, CD31 immunostaining confirmed a significant increase in vessel density in the CS-derived tumors at the 8-week observation time point compared with ICS-derived tumors.
In summary, we have designed and characterized a new, preclinical ovarian cancer animal model utilizing an immunocompetent host and CS technology to form orthotropic, syngeneic allografts with properties that closely replicate human EOC. We have also demonstrated advantages of this model, including the potential benefits directly related to CS technology (intact cell junctions, surface protein and growth factor expression, enhanced angiogenesis, accelerated initiation, growth, invasion, and metastases) as well as the advantages of orthotopic engraftment, which more closely mimics the origin of human cancer. We hypothesize that this model may serve as a more accurate predictor of future clinical efficacy and provide for more economical screening of anticancer drug candidates in the future.
Footnotes
Acknowledgments
The authors thank Yongen Sun, MD, for his expertise and surgical skills in conducting animal experiments. This study was supported by grants from the University of Utah's Department of Obstetrics and Gynecology, the University of Utah Program in Personalized Health Care and Formation of Innovation Center for Fusion of Advanced Technologies in the Special Coordination Funds for Promoting Science and Technology “Cell Sheet Tissue Engineering Center (CSTEC),” and the Global Center of Excellence (GCOE) Program, Multidisciplinary Education and Technology and Research Center for Regenerative Medicine (MERCREM), from the Ministry of Education, Culture, Sports Science, and Technology (MEXST), Japan.
Disclosure Statement
Teruo Okano is a founder and director of the board of CellSeed, Inc., licensing technologies and patents from Tokyo Women's Medical University. Teruo Okano is also a shareholder of CellSeed, Inc., Tokyo Women's Medical University and is receiving research funds from CellSeed, Inc.