
Abstract
Patterned three-dimensional (3D) cell culture models aim to more accurately represent the in vivo architecture of a tissue for the purposes of testing drugs, studying multicellular biology, or engineering functional tissues. However, patterning 3D multicellular structures within very soft hydrogels (<500 Pa) that mimic the physicochemical environment of many tissues remains a challenge for existing methods. To overcome this challenge, we use a Sacrificial Micromolding technique to temporarily form spatially and geometrically defined 3D cell aggregates in degradable scaffolds before transferring and culturing them in a reconstituted extracellular matrix. Herein, we demonstrate that Sacrificial Micromolding (1) promotes cyst formation and proper polarization of established epithelial cell lines, (2) allows reconstitution of heterotypic cell–cell interactions in multicomponent epithelia, and (3) can be used to control the lumenization-state of epithelial cysts as a function of tissue size. In addition, we discuss the potential of Sacrificial Micromolding as a cell-patterning tool for future studies.
Introduction
I
A common strategy for controlling tissue geometry in 3D culture is the use of photolithographic techniques for patterning or replica-molding scaffolds that can accommodate collections of cells at high density and with a well-defined geometry.5,6 The materials typically used in this approach are those that can be readily patterned by photolithography (e.g., epoxy-based photoresists and UV-reactive polymers) or micromolded by soft lithography (e.g., elastomers and agarose). For example, polydimethylsiloxane (PDMS), agarose, or poly (ethylene glycol) (PEG) microwells provide semi-3D (or 2.5D) cell culture platforms that produce complex tissue geometries at low cost and high throughput.7–9 Unfortunately, these materials do not recapitulate the physicochemical properties of the in vivo extracellular matrix (ECM). Moreover, these substrates are generally limited to 3D extrusions of two-dimensional (2D) patterns and do not fully encapsulate the reconstituted tissue. Therefore, elaboration of these photolithographic approaches allowed the formation of spatially and geometrically defined 3D cellular patterns within biomimetic ECM gels. For example, Nelson et al. used an elastomeric array of well-defined posts to imprint microcavities within type I collagen gels. 10 When seeded with cells and overlaid with additional collagen, this method offered an unprecedented level of control over the geometry and spatial organization of 3D tissues within a reconstituted ECM. Nevertheless, this strategy is challenging to implement when dealing with very soft gels such as the popular, laminin-rich, reconstituted basement membrane hydrogel known as the Matrigel.
Matrigel is unparalleled in its emulation of the basement membrane in vivo, due to a multifactorial composition of distinct matrix components (laminin, collagen IV, entactin, etc.) as well as its mechanical properties that are well matched to many soft tissues in vivo.11,12 Unlike many other ECM models, Matrigel forms stable but extremely soft gels (<0.5 kPa) 13 that promotes morphogenesis and allows the diffusion of biochemical factors throughout the 3D matrix. However, the same liquid-like time-dependent viscoelastic properties that define the gel's low stiffness also provide a challenge for the reliable geometric patterning of this soft material. Toward overcoming this challenge, Sodunke et al. used a combination of lithographic and molding techniques to pattern freestanding circular or rectangular islands of Matrigel on a glass substrate. 14 In this method, the concentration of cells within the Matrigel islands was controlled by predefining the dilution of cells suspended in the liquid Matrigel molded against structural supports made out of PDMS or polyHEMA. Therefore, the authors were able to pattern Matrigel islands containing a single cell that grows into a single tissue, on average. However, this method relies on Poisson statistics for loading the islands. As a consequence, many islands contain more than one cell or remain empty. Moreover, this strategy does not provide geometric and spatial control of multicellular architectures fully embedded within a soft gel. We therefore sought an improved micromolding strategy for patterning cells into spatially and geometrically controlled multicellular structures fully embedded in Matrigel or other soft gels.
Inspired by previous reports that used degradable materials such as gelatin for patterning microfluidic channels embedded in collagen or fibrin,15,16 we envisioned using this material as a degradable scaffold for micromolding spatially and geometrically resolved multicellular structures that can be transferred to soft biomimetic gels. In this study, we identify conditions that preserve the geometry and spatial location of multicellular structures transferred to Matrigel or fibrin after gelatin degradation. We also demonstrate that tissues formed using this Sacrificial Micromolding strategy are morphologically normal and are amenable to long-term culture and imaging.
Materials and Methods
Cell lines in 2D culture conditions
MCF10A, MDCK, and Caco2 cells were maintained in 2D cell cultures as previously described.17,18 Primary human mammary epithelial cells (HMEC, specimen 240L) were derived from the cosmetic reduction mammoplasty of human patients and maintained in 2D cell culture for four passages in the M87A medium before use in experiments.7,19 All primary human cells were provided by Dr. Martha Stampfer (Lawrence Berkeley National Laboratory) as deidentified human specimens.
Fabrication of sacrificial microwells
Freestanding SU-8 features on silicon wafers were fabricated using photolithographic techniques. Photomasks were designed in AutoCAD and printed by Outputcity Co. Protocols for photopatterning were adapted from MicroChem's technical specification sheets. For example, circular microwells 120 μm in diameter and 80 μm deep were fabricated using SU-8 2035 (MicroChem) spun onto silicon wafers at a speed of 500 rpm for 10 s followed by a 1250 rpm spin for 30 s. The wafer was then soft baked for 5 min at 65°C, for 10 min at 95°C, and UV exposure in contact mode at an energy of 215 mJ/cm2. Wafers were then postexposure baked for 5 min at 65°C and for 10 min at 95°C, and developed in the SU-8 developer (MicroChem) for at least 20 min. The patterned substrate was then washed with isopropanol/water and hard baked at 150°C for 1 h before measuring pillar heights using a stylus profilometer (Ambios XP2) and visualizing the patterned wafer by SEM (NovelX mySEM). This silicon master was used to create PDMS micropillars by pouring a Sylgard 184 (Silicone Elastomer Kit; Dow Corning) onto the patterned wafer using a base:crosslinker ratio of 10:1. After overnight incubation at 65°C, the cured PDMS template was peeled off the silicon master, cut into small rectangular stamps that fit the well of a 24-well plate, and incubated with Sigmacote (Sl-2; Sigma-Aldrich) for several hours to ease the release of the PDMS from the protein-based sacrificial layer.10,20 Supplementary Figure S1 (Supplementary Data are available online at
Sacrificial micromolding
Sacrificial microwells were molded from a solution of 8% (w/v) gelatin (Knox Gelatine) in phosphate-buffered saline (PBS). This sacrificial gelatin mold was generally formed within the wells of a 24-well plate by depositing the PDMS stamp onto a 250 μL drop of liquid gelatin. Gelatin gelation occurred upon placing the 24-well plate at 4°C for 30 min or longer. To form aggregates, cells were dissociated from culture plates, resuspended in 0.5 mL of media at a concentration of 106/mL, pipetted onto the imprinted gelatin mold in the 24-well plate, and centrifuged into the sacrificial microwells at 160 g at 4°C for 4 min immediately after PDMS removal. Excess cells were then washed away with a medium. To transfer the remaining physically confined cell aggregates from gelatin to Matrigel, we first removed the excess gelatin from the unpatterned regions surrounding the sacrificial microwells using a sterile spatula. We then carefully peeled the (∼200–300 μm thick) gelatin mold with cell clusters from the substrate and inverted it over a 120 μL Matrigel slab (previously set for 30 min at 37°C within the well of a 24-well plate or other vessels), incubated the dish at 37°C for 1 h, gently washed away the melted gelatin with warm media, and finally overlaid an additional 120 μL of Matrigel on top before adding the media for long-term culture. Figure 1A and B offer a schematic representation of the Sacrificial Micromolding procedure.
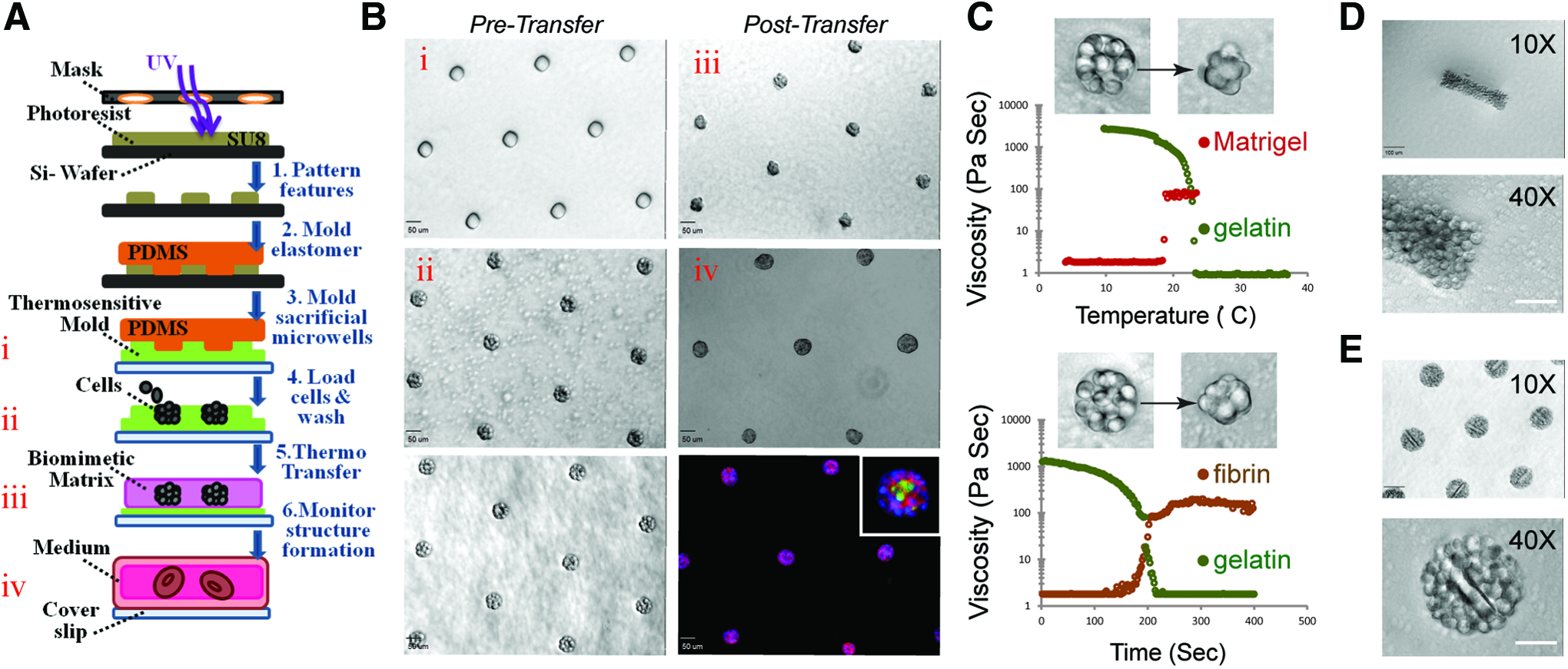
Sacrificial Micromolding of three-dimensional (3D) tissues in soft gels.
Cell count and cell viability
To estimate the number of cells within the small spherical clusters assembled within the gelatin microwells fabricated using PDMS posts 60 μm in diameter and 80 μm tall, we fixed and stained the nuclei of MDCK cells immediately after transfer to Matrigel as described in the Immunofluorescence section. Then, we manually counted the number of intact nuclei for 30 cell aggregates using 3D confocal reconstructions of the tissue. For the larger cell aggregates prepared using PDMS posts 120 or 240 μm in diameter and 80 μm tall, the exact number of nuclei within the tissue could not be easily resolved by confocal microscopy. Therefore, we decided to classify the dimensions of different spherical cell aggregates by measuring the diameter of the reconstituted tissue through image analysis. Cell viability of reconstituted cell aggregates was accessed by staining microtissues with Trypan blue immediately after transferring them from gelatin to poly-lysine-coated glass slides.
Formation of heterotypic cell aggregates
Human mammary luminal (LEP) and myoepithelial cells (MEP) were fluorescently activated cell sorting purified from fourth passage primary HMEC using established cell surface markers (CD10 for MEP and CD227 for LEP), as previously described. 7 To form heterotypic aggregates, MEP and LEP were resuspended at a concentration of 106/mL and mixed at a 1:1 ratio immediately before loading the sacrificial microwells.
Tracking cell lineages within heterotypic cell aggregates
To track the position of MEP and LEP as a function of time, we stained these cells with live-cell cytosolic tracking dyes immediately before loading into the sacrificial gelatin mold. The staining procedure was carried out following the manufacturer's instructions and required resuspending sorted LEP or MEP in 10 mL of PBS with a final concentration of 1 μM of either CellTracker Green (Invitrogen) for LEP or 1 μM CellTracker Red (Invitrogen) for MEP for 5 min at 37°C. The stained cells were then pelleted and resuspended at the desired concentration before micromolding (i.e., 106/mL).
Fabrication of polymeric microstructures
PEG-based polymeric microrods were fabricated using photolithographic techniques, as previously described. 21 To incorporate these microstructures within reconstituted cell aggregates, we mixed the (100×15×15 μm) microrods with MDCK cells at a ratio of 1:100 and loaded the mixture into sacrificial gelatin microwells molded from PDMS posts 120 μm in diameter and 80 μm in height.
Characterization of hydrogels
The viscosity of Matrigel, fibrin, and gelatin was measured as a function of temperature and/or time. Matrigel (Lot 07898; BD Biosciences 354230) was used undiluted. Fibrin gels were made by mixing a solution of 5 mg/mL fibrinogen (Sigma F8630) and 0.1 U/mL thrombin (Sigma T7513) in PBS. Gelatin (Knox ORIGINAL Gelatine) was purchased at Safeway and bloomed to 8% w/v in cold PBS before boiling. All viscosity measurements were accomplished using a BlackPearl Viscometer (ATS Rheosystems) and a 30-mm parallel plate (with 1 mm gap) insert (ATS Rheosystems). For the thermal sweeps of Matrigel and gelatin, we measured gel viscosity as a function of increasing (for Matrigel) or decreasing (for gelatin) temperature. For the time sweeps of fibrin and gelatin, we measured viscosity as a function of time at constant temperature (37°C). All parameters used for captured measurements were controlled using Rheosys Micra software (ATS Rheosystems).
Immunofluorescence
All microtissues were fixed with 4% formaldehyde (in PBS) for at least 20 min and then incubated in a blocking buffer (10% heat-inactivated goat serum in PBS+0.5% Triton X-100) at 4°C for at least 1 day to guarantee the diffusion of the higher molecular weight proteins to the core of the reconstituted microtissues. Primary antibodies were diluted in the blocking buffer and then added to the sample, as summarized in Table 1. After at least 1 day incubating at 4°C with the primary antibody, all microtissues were washed several times with PBS+0.05% Triton X for at least 1 day and incubated with Alexafluor-conjugated secondary antibodies (Life Technologies) diluted at a concentration of 1:200 in the blocking buffer for ∼1 day. Before imaging the clusters, all samples were washed with PBS+0.05% Triton X+1 μg/mL DAPI for at least 1 day.
The dilution used for each application is reported in parentheses.
FACS, fluorescently activated cell sorting; IF, immunofluorescence.
Image acquisition
All confocal microscopy images were acquired using an inverted confocal microscope (Zeiss Cell Observer Z1) equipped with a Yokagawa spinning disk, an Evolve EM-CCD camera (Photometrics), and running Zeiss Zen Software. All other images were acquired using an inverted epifluoresence microscope (Zeiss Axiovert 200M) running SlideBook software.
Quantification of keratin localization
For quantifying the localization of keratin markers (i.e., K19 for LEP and K14 for MEP) within reconstituted HMEC microtissues, we collected 20 confocal (40× magnification) images of 20 fixed and stained structures, cropped the images to the exact size of the spherical tissue, binarized the fluorescent signal from the green (for K19) and red (for K14) channels, stacked/resized to the smallest sample of all images collected, and plotted the normalized average radial reslice of the normalized average (K14 and K19) signals. Image analysis was performed using FIJI software.
Quantification of lumenization-state
For quantifying the number of lumens formed as a function of the diameter of the reconstituted microtissue, we first measured the diameter of 30 microtissues in FIJI and plotted the average and standard deviation of our measurements against the known diameter of the PDMS post. We then visually counted the number of lumens formed as determined by Z-stack confocal analysis of 11 MDCK tissues (for each size), fixed and stained with phalloidin and DAPI, and plotted the average of the number of lumens as a function of the average diameter of the tissue. Lumens were defined as phalloidin-stained actin rings surrounded by DAPI-stained nuclei.
Results
Sacrificial Micromolding and phase transitions of gelatin
Gelatin was selected for Sacrificial Micromolding because it is a nontoxic hydrogel that can be micromolded at 4°C and liquefied at 37°C. As shown in Figure 1C, when a 1 mm slab of cooled gelatin was incubated at 37°C, its viscosity dropped dramatically as measured by viscometry. On the other hand, the viscosity of a liquid drop of Matrigel sharply increased as the gel was brought toward the same physiological temperature. We therefore reasoned that degradable gelatin microwells could be used to control the initial geometry and location of multicellular aggregates in 3D without significantly affecting cell viability upon degradation (Fig. 1A and Supplementary Fig. S1). We used this material to pattern a hexagonal array of spherical microtissues of ∼13±3 (standard deviation) MDCK cells in Matrigel before probing for protein localization through immunofluorescence after 4 days in culture at 37°C (Fig. 1B). Rectangular cell aggregates were also prepared by simply varying the geometric dimensions of the PDMS stamps used to imprint the sacrificial microcavities (Fig. 1D). Polymeric microstructures such as PEG-based microrods were incorporated within predefined multicellular architectures when mixed with cell suspension before Sacrificial Micromolding (Fig. 1E). Furthermore, Sacrificial Micromolding was also used to encapsulate microtissues in other soft gels like fibrin—a biomimetic hydrogel that exhibits controllable liquid–gel transitions upon the addition of thrombin to the fibronectin precursor (Fig. 1C, bottom).
Patterning MDCK, MCF10A, and Caco2 cysts by Sacrificial Micromolding
To validate the use of Sacrificial Micromolding for 3D tissue culture, we investigated whether well-characterized epithelial cell lines known to form hollow cysts when cultured in Matrigel could recapitulate their established polarized tissue architecture when transferred from gelatin microwells to Matrigel.22–24 We found that after only 4 days, all cell lines established polarity and features indicative of cyst formation (Fig. 2). GP135 and actin were found lining the lumen of MDCK and Caco2 microtissues, respectively (Fig. 1B bottom right insert and Fig. 2B bottom right). Consistent with previous reports, 22 MCF10A cells did not establish true apical–basal polarity in Matrigel (Fig. 2B top right). However, we observed proper localization of α6 Integrin at the basal surface and E-cadherin to cell–cell junctions indicating that MCF10A microtissues established tissue-level polarity.
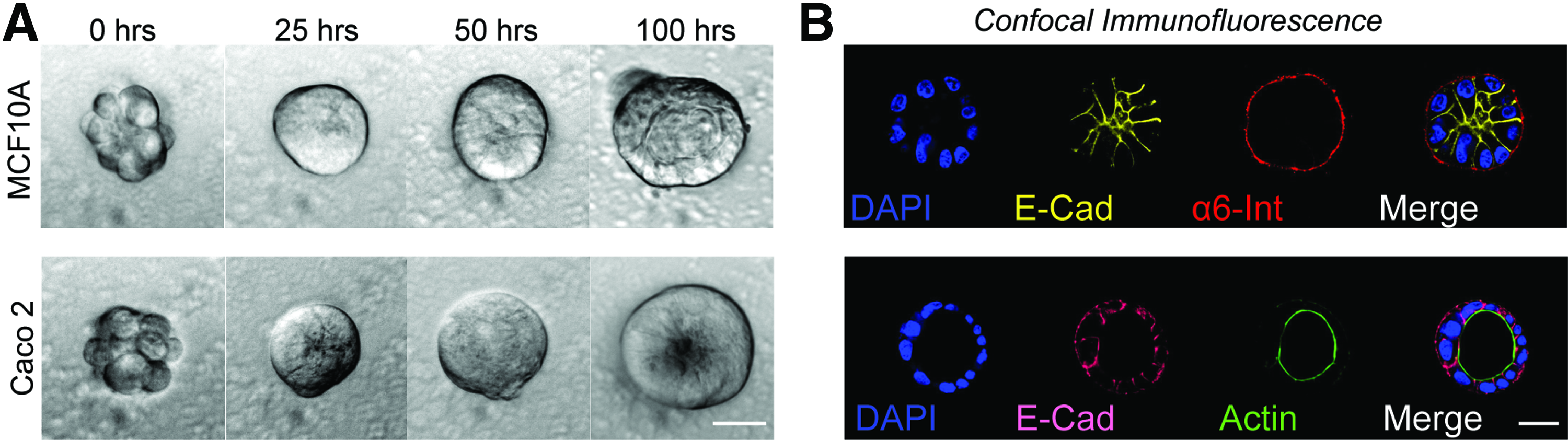
Polarization and lumen formation of epithelial cell lines.
Formation of properly organized heterotypic mammary epithelial tissues by Sacrificial Micromolding
Cell-patterning technologies can define the initial spatial position of cells in a gel, but shortly after tissue reconstitution, cells can self-organize according to a balance of cell-intrinsic and cell-extrinsic cues. 7 We therefore tested whether Sacrificial Micromolding would support the proper self-organization of the two principal cell types found in the human mammary epithelium by providing the appropriate extrinsic cues. Encouragingly, we found that heterogeneous mixtures of luminal and myoepithelial cells self-organized and maintained architecture after 2 days in Matrigel (Fig. 3). We quantified the extent of organization in these tissues utilizing immunofluorescence to characterize the average spatial localization of lineage-specific keratin markers at 1 h (Fig. 3C) and 48 h (Fig. 3D) after tissue reconstitution. Consistent with the in vivo tissue organization, we reproducibly found luminal (LEP: K14−/K19+) epithelial cells in the core surrounded by an outer layer of myoepithelial (MEP: K14+/K19−) cells. Therefore, Sacrificial Micromolding and transfer to Matrigel provide an efficient means of forming and culturing primary human MEP and LEP into tissues that mimic the in vivo architecture of the mammary gland. In contrast, formation of identical cell aggregates in PDMS microwells was not a suitable method for long-term culture. In PDMS microwells, MEP began to spread over the stiff and adhesive 2D interface, and the compacted architecture was lost when cell aggregates interacted with the elastomer for more than 24 h (Fig. 3B).

Self-organization of primary human mammary epithelial cells (HMEC) in Matrigel.
Lumenization as a function of tissue size
One of the advantages of using Sacrificial Micromolding for 3D tissue formation and culture is the precise control over the initial size of reconstituted cell aggregates. We formed MDCK cell aggregates in gelatin wells of different sizes and then cultured them for 4 days in Matrigel. Sacrifical Micromolding reproducibly defined the average diameter of the reconstituted cell aggregates in 3D (Fig. 4). Therefore, we used this feature of the technique to quantify the average number of lumens formed as a function of microtissue diameter at fixed time points after aggregation (Fig. 4C). Previous studies indicate that lumen formation in MDCK occurs spontaneously at the interface between groups of cells in contact with the ECM. 25 When multiple lumen form in a tissue, they can coalesce as morphogenesis progresses. However, the precise relationship between the tissue size and lumen formation has not been established. We therefore formed cell aggregates incorporating ranging from 50 to 200 μm in diameter by Sacrificial Micromolding before allowing them to grow in 3D culture. At 4 days, we noticed that the number of lumens in each tissue was a function of the diameter of the initial aggregate (Fig. 4C). Interestingly, many of the lumen formed between aggregates of cells not in direct contact with the surrounding Matrigel. Future studies of multicellular cyst formation as a function of time and geometry will help to elucidate how these multicellular constructs regulate lumen formation and coalescence.
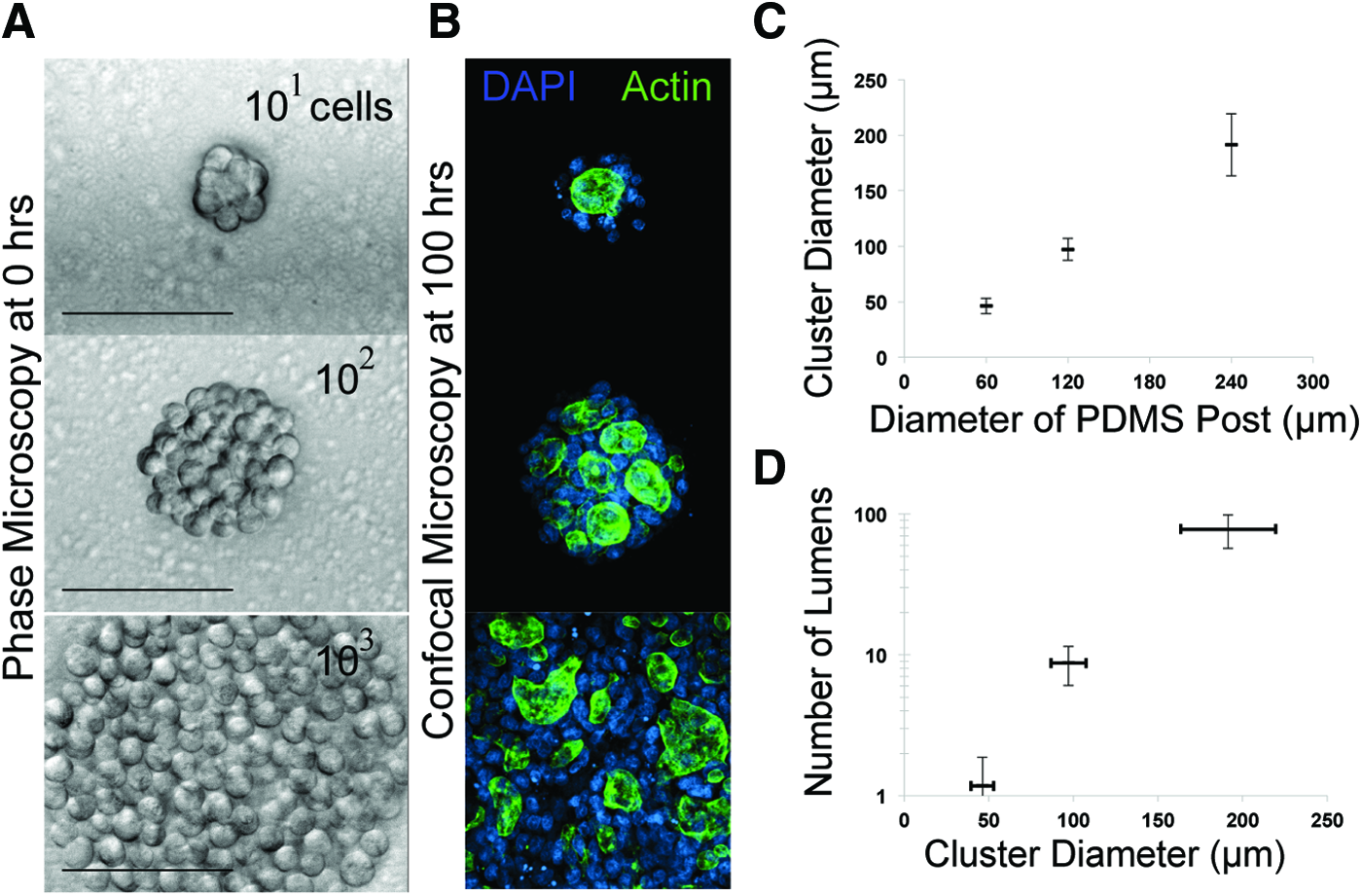
Quantifying the number of lumens formed as a function of aggregate size.
Discussion
Tissues demonstrate a high degree of architectural complexity in vivo. The capacity to effectively and stably reproduce these structures in vitro is critical for the study and engineering of mammalian tissues. We take advantage of the phase-transition properties of gelatin to precisely define the initial size, geometry, and location of reconstituted tissues fully embedded in very soft gels like Matrigel and fibrin (Fig. 1). We find that when small spherical multicellular structures of MDCK, MCF10A, or Caco2 cells with an approximate diameter of 50 μm were cultured in Matrigel, the reconstituted cell aggregates formed properly polarized microtissues containing a single lumen after only 4 days (Fig. 2)—a process that may take longer when starting from single cells embedded in Matrigel. Furthermore, we were able to reconstitute tissues in the presence of microparticles and to carefully tune the dimensions of epithelial cell aggregates to control the number of lumens formed within each tissue (Figs. 1 and 3). Consistent with the notion that Sacrificial Micromolding creates a proper tissue microenvironment in 3D culture, aggregates of primary human mammary luminal and myoepithelial cells self-organized properly after transfer to Matrigel. Moreover, these results indicate that Sacrificial Micromolding is also capable of recapitulating heterotypic cellular interactions and the essential processes of self-organization necessary to support the morphogenesis of more complex tissues. However, further manipulation of gel formulation will be required to identify the optimal physicochemical properties necessary to promote distinct aspects of morphogenesis characteristic of each tissue type. For example, addition of low concentrations of collagen or increasing gel stiffness with synthetic materials could promote cell dissemination or branching morphogenesis.26,27 Given the flexibility of the technique, we anticipate that Sacrificial Micromolding will find utility in the study of diverse heterotypic cellular interactions characteristic of all metazoan organisms.
For most of our experiments, we use Matrigel as a laminin-rich ECM because it is widely used to mimic the physicochemical composition of the basement membrane enveloping all epithelial tissues in vivo. However, this gel is derived from a mouse sarcoma and exhibits high compositional variability among different commercial lot numbers. For these and other reasons, tissues that are reconstituted in Matrigel are unlikely to find clinical applications and are currently limited to in vitro explorations of tissue biology. Despite this limitation of Matrigel, we expect that this method can be extended to encapsulate multicellular aggregates within a spectrum of other hydrogel-based scaffolds currently under development to bypass the molecular complexity and compositional variability of Matrigel. 28
Footnotes
Acknowledgments
AC was supported by the U.S. Department of Defense through the NDSEG program. The CDMRP (W81XWH-10-1-1023 and W81XWH-13-1-0221), the UCSF Center for Systems and Synthetic Biology (P50 GM081879), the Program in Biomedical Breakthrough Research (PBBR), and the NIH (DP2 HD080351-01) also contributed to this research. The authors also thank the BMNC at UCSF for training and use of their photolithographic facilities.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.