
Abstract
In vitro coculture models mimicking the bronchial barrier are a significant step forward in investigating the behavior and function of the upper respiratory tract mucosa. To date, mostly synthetic materials have been used as substrates to culture the cells. However, decellularized tissues provide a more in vivo-like environment based on the native extracellular matrix. In this study, an in vitro, bronchial wall coculture model has been established using a decellularized, porcine luminal trachea membrane and employing three relevant human cell types. The tissue was decellularized and placed in plastic transwell supports. The human bronchial epithelial cell line, 16HBE14o-, was seeded on the apical side of the membrane with the human lung fibroblast cell line, Wi-38, and/or the microvascular endothelial cell line, ISO-HAS-1, seeded on the basolateral side. Transepithelial electrical resistance (TER) was measured over 10 days and tight/adherens junctions (ZO-1, occludin/β-catenin) were studied through immunofluorescence. Scanning electron microscopy (SEM) was performed to evaluate microvilli and cilia formation. All cultures grew successfully on the membrane. TER values of 555 Ω·cm2 (±21, SEM) were achieved in the monoculture. Cocultures with fibroblasts reached 565 Ω·cm2 (±41, SEM), with endothelial cells at 638 Ω·cm2 (±37, SEM), and the triple culture achieved 552 Ω·cm2 (±38, SEM). ZO-1, occludin, and β-catenin were expressed in 16HBE14o- under all culture conditions. Using SEM, a dense microvilli population was found. Prominent cell–cell contacts and clusters of emerging cilia could be identified. Fibroblasts and endothelial cells strengthened the formation of a tight barrier by the 16HBE14o-. Thus, the coculture of three relevant cell types in combination with native decellularized scaffolds as a substrate approaches more closely the in vivo situation and could be used to study mechanisms of upper respiratory damage and regeneration.
Introduction
A
The mucosa of the airway exerts an important function in the protection against inhaled drugs and compounds, including particulate matter. Epithelial cells form a barrier that blocks the impact of external irritants, preventing their entry into the deeper tissues, but still allowing the diffusion of water, soluble ions, and immune cells.9,10 To understand better the mechanisms involved in the formation and maintenance of this barrier, some in vitro coculture models have been developed.11–14 These cocultures, consisting of epithelial cells, endothelial cells, or fibroblasts, have not only allowed the study of intercellular interaction and communication but have also been shown to be more efficient in the construction of the barrier than monocultured bronchial cells.12,13
The barrier function of the epithelium is provided by tight cell–cell contacts (tight junctions [TJs] and adherens junctions). With their help, the cells form a continuous monolayer that seals the body from the external environment. These junctions are also responsible for the regulation of pericellular diffusion and participate in the maintenance of cell polarity.15,16 The formation of TJs can be monitored through the establishment of a transepithelial electrical resistance (TER), which can be measured in living cells.17,18
The epithelial barrier is a highly dynamic entity organized to fulfill specific physiological functions, which can be affected by many external factors. These include cellular cross talk with neighboring cells, growth and differentiation hormones, pharmacological agents, cell cycle stages, and the composition of the ECM.15,19 Current research is aimed at establishing multicellular coculture models to investigate the above-mentioned physiological functions12,20 in a more realistic in vivo-like manner. Several on-going coculture studies use synthetic or artificial cell culture substrates. However, the composition, stiffness, and elasticity of the ECM plays a crucial role in cell behavior, thus influencing cell adhesion, proliferation, and differentiation,21–25 as well as the recellularization process on acellular scaffolds.26,27 To approach more closely the in vivo situation, the next step would be a multicellular coculture comprising several tissue-relevant cell types in combination with a native decellularized scaffold as a substrate. This in vitro tissue construct could be used to allow more realistic studies on mechanisms of upper respiratory damage and regeneration.
For this reason, we have developed and characterized in the present study a transwell system that comprises a membrane made of decellularized tracheal tissue. The latter provided a substrate formed by the mucosa and submucosa of the pig trachea, containing the ECM on which bronchial epithelial cells would grow naturally. The biocompatibility of this membrane was assessed and compared with commercial polycarbonate transwells. A bronchial epithelial cell line, 16HBE14o-, was grown in monoculture on one side of the membrane or in coculture with two further relevant cell types, namely the fibroblast cell line, Wi-38, and the microvascular endothelial cell line, ISO-HAS-1, which have been previously used in transwell studies.11,12,28,29 The bronchial epithelial cells were grown to confluence and examined for the development of cell–cell junctions. Barrier formation was evaluated by measuring TER. Scanning electron microscopy (SEM) was also used to determine the presence of microvilli and cilia on the luminal surface of the epithelial cells and simultaneously permitted detection of the fibroblasts and endothelial cells growing in the coculture conditions.
Materials and Methods
Trachea decellularization
Tracheal tissue excised from 40 healthy pigs was obtained from a local slaughterhouse. The entire trachea was isolated and washed with phosphate-buffered saline (PBS) containing antibiotics (streptomycin at 90 μg/mL, penicillin at 50 U/mL) and antimycotics (amphotericin [25 μg/mL]). To enhance cell disruption, the tissue was frozen at −80°C for 30 min and immediately thawed in a water bath at 37°C. This freeze–thaw cycle was repeated four times. Subsequently, the trachea was submerged for 48 h in deionized water, followed by rinsing several times with PBS. This and the following steps were performed under constant agitation at 4°C. To eliminate the cell debris from the tissue, the tracheas were submerged in a solution that contained 8 mM of 3-[(3-cholamidopropyl) dimethylammonio]-1-propanesulfonate (CHAPS), 1M NaCl, and 25 mM EDTA in PBS, as previously described. 30 This was performed during a period of 5 days, exchanging the CHAPS solution twice daily. The tissue was then rinsed in PBS (containing antibiotics and antimycotics) and washed for 7 days, exchanging the PBS solution daily. After this washing step, the tunica mucosa and submucosa (the luminal side of the tracheas), which form a membrane-like structure, were excised and used for the experiments as a membrane equivalent to the filter membrane of the conventional transwell system. The fibroelastic layer and tunica adventitia below were discarded. The decellularized membrane was then either processed for staining and SEM evaluation or assembled in a plastic cellcrown (Scaffdex) to form the biotranswell. The apical side of the membrane (epithelial side) was oriented to form the top surface of the biotranswell and corresponded to the tunica mucosa. The lower surface was formed by the basolateral aspect (tunica submucosa) of the membrane (Fig. 1A).
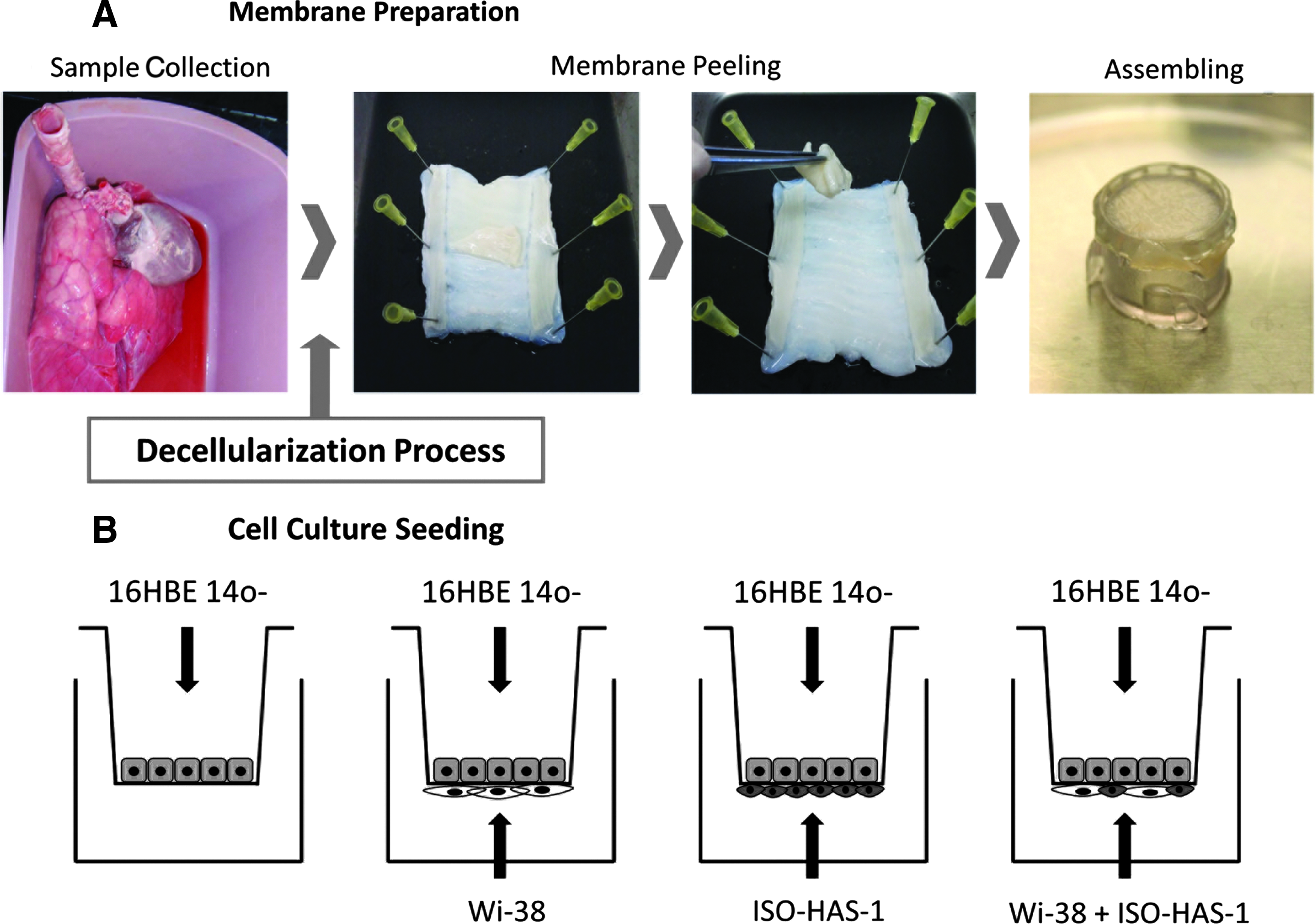
The biotranswell.
Immunofluorescence staining for ECM proteins
Segments of ∼1 cm2 were embedded in optimal cutting temperature compound (OCT; Sakura) and snap-frozen in liquid nitrogen. The blocks were sectioned in a cryostat and the 10-μm sections were placed on top of coverslips, which were previously functionalized, by incubation for 5 min in a solution of (3-aminopropyl) trimethoxy-silane 2% in acetone, followed by rinsing twice in acetone for 5 min and washing with deionized water.
Once the sections were obtained, the coverslips were rinsed with PBS several times to remove the OCT. Samples were blocked with a solution containing 1×TBS, 0.5% Triton X-100, 6% fetal bovine serum (FBS), and 1% bovine serum albumin (BSA). A primary antibody against laminin (1:20; Sigma) was incubated in a solution of 1×TBS, 0.1% Triton X-100, 6% FBS, and 0.1% BSA overnight at 4°C. Samples were allowed to warm up and rinsed thrice for 10 min with the same solution used for the primary antibody incubation. Secondary donkey anti-rabbit Cy3 (1:200; Jackson) antibody was incubated in a solution of 1×TBS, 0.1% Triton X-100, 6% FBS serum, and 0.1% BSA for 2 h. The secondary antibody was then rinsed from the sample, performing three washing steps (15 min each) with TBS 1×.
Alcian Blue and van Gieson staining
Samples of decellularized tissue (for ECM characterization) were dehydrated with ethanol gradients, embedded in paraffin, sectioned on a microtome (7 μm thickness), and deparaffinized in xylene. Following rehydration in graded alcohols, the sections were stained following a classical protocol either for van Gieson or alcian blue staining. Samples stained with alcian blue were also counterstained with hematoxylin so that the nuclei could be detected. Sections were observed and imaged in a conventional bright-field microscope (TE2000; Nikon).
Confocal microscopy and multiphoton microscopy (two-photon microscopy and second harmonic generation microscopy)
Before imaging, the tracheal sections were stained with Hoechst dye (HOE 33342; Sigma) to allow visualization of any remaining nuclei. The wavelengths used in the two-photon microscopy allowed a visualization of nuclei and elastin using the same channel. 31 Collagen and elastin were imaged using a nonlinear technique of second harmonic generation (SHG) and two-photon excited fluorescence (TPEF) in a simultaneous mode.32,33 Collagen has a highly crystalline triple-helix structure that is noncentrosymmetric. This makes it very efficient in generating the second harmonic of incident light. 34 Elastin, normally present along with collagen, is a significant source of ECM autofluorescence and can be imaged with TPEF. 34 The experimental arrangement was based on a Leica inverted confocal laser scanning microscope SP-5 with an META scanning module equipped with a mode-locked Ti:sapphire femtosecond laser. In this study, the laser emission was 810 nm, and two channels were used to simultaneously detect the SHG and TPEF signals. The detection spectral range was a narrow band (387–409) for the SHG signal of the collagen and a broad band (450–600 nm) for the TPEF signal associated with the elastic fibers. The images of the 12-μm sections were collected using a Leica 25×0.95-NA water objective. Images were taken with 1024×1024 pixels and had a 16-bit pixel depth. The step size of the z-stacks was of 2.1 μm. For the multiphoton techniques, the average laser powers were 4 mW in the TPEF detection channel and 7 mW in the SHG detection channel, respectively.
On a separate channel, laminin was imaged by switching the system to a normal confocal mode. The final image comprised three channels, two for collagen and elastin that had been acquired by multiphoton microscopy and one that originated from laminin, imaged by confocal microscopy.
Scanning electron microscopy
Samples for SEM were prepared using standard procedures with fixation using 2% glutaraldehyde and 2.5% paraformaldehyde in 0.1 M cacodylate buffer (EMD Biosciences) for 1 h at room temperature, then rinsing in cacodylate buffer, and dehydration in an ethanol gradient. Samples were further dehydrated in hexamethyldisilizane for 10 min and dried overnight, then sputter coated with gold and analyzed using a JEOL-6510 or a JEOL-7100.
Cell culture
Three different cell lines were used in these studies: bronchial epithelial cells, 16HBE14o-, obtained from Dr. Gruenert (University of California, Oakland, CA), human embryonic lung fibroblasts, Wi-38 (ATCC-CCL-75), and the angiosarcoma cell line, ISO-HAS-1, derived from a single clone of the hemangiosarcoma cell line, ISO-HAS, previously described.35,36 Cells were maintained in culture with Eagle's minimal essential medium supplemented with 10% fetal calf serum (FCS), 1% penicillin/streptomycin, 2% sodium bicarbonate, 1% Glutamax, and 1% sodium pyruvate using standard cell culture methods. Both the medium and supplements were purchased from Sigma. ISO-HAS-1 and 16HBE14o- cells were split at a ratio of 1:5 and Wi-38 fibroblasts at 1:3. For all cell types, passages 10–30 were used in the current studies.
Cell seeding
Bronchial epithelial cells (16HBE14o-) were cultured alone or with fibroblasts (Wi-38) and/or endothelial cells (ISO-HAS-1). To form the biotranswell, the decellularized trachea membrane was placed in a plastic cellcrown (Scaffdex) (Fig. 1A). 16HBE14o- were seeded on the top side of a biotranswell and a commercial HTS 12-Transwell filter (polycarbonate, 0.4 μm pore size, 3513; Costar), as depicted in Figure 1B. For the cultures on the polycarbonate filter, the upper and lower surfaces of the transwells were coated with a solution of collagen type 1 in 0.1 M acetic acid and air-dried at room temperature before the addition of cells. The biotranswells were checked for leakage by adding 0.5 mL of medium to the upper compartment and observing the lower compartment for the appearance of the medium during a 10-min period.
The human fibroblast cell line, Wi-38 (3×104 cells/cm2), the human endothelial cell line, ISO-HAS-1 (5×104 cells/cm2), and a mixture of both cell lines (at the same concentration, respectively) were placed on the lower surfaces of inverted polycarbonate transwell filter membranes or the biotranswells and incubated for 2 h at 37°C and 5% CO2 (Fig. 1B).
All filters were then inverted and placed in 12-well plates filled with 1.5 mL of medium per well. The bronchial cells, 16HBE14o-, were then seeded on the top surface of the filters (2.5×105 cells/cm2) in a volume of 500 μL medium. The cultures were grown simultaneously with the corresponding cell type on the lower side or alone (monocultures) during a period of 8 days on the polycarbonate membranes and during a period of 12 days on the biotranswells. The medium was exchanged every 2 days.
Measurement of TER or bioelectrical measurements
Electrical resistance across the monolayer and bilayer was measured using an EVOM voltohmmeter (World Precision Instruments) with STX-2 chopstick electrodes. The TER was measured every 2 days throughout the experiment.
The resistance from the blank value obtained from the polycarbonate transwell or biotranswell was subtracted from each well to calculate the resistance exerted by the cell layer. Subsequently, the value was multiplied by the area of the membrane to obtain the corresponding Ω·cm2 value. Resistance was shown as the mean resistance×area of at least two independent experiments (n=5–10 in the biotranswells and n=6 in the polycarbonate transwells for each experiment).
Immunofluorescence staining of the cell cultures
After culturing the cells, the different membranes were fixed with 3.7% paraformaldehyde for 15 min and then washed thrice with PBS. Cells were permeabilized and blocked during a period of 30 min with a solution based on TBS, 0.5% Triton X-100, 6% FBS, and 1% BSA. Then, primary antibodies against ZO-1 (1:200, 61-7300, Zymed), β-catenin (1:200, 610154; BD), vimentin (1:200, M 7020; Dako), and CD31 (1:50, M 0823; Dako) were incubated in a solution of 1×TBS, 0.1% Triton X-100, 6% FBS, and 0.1% BSA overnight at 4°C. Samples were allowed to warm up and were rinsed thrice for 20 min with the same solution, in which the primary antibody had been diluted. Secondary antibodies (donkey anti-rabbit Alexa 488; donkey anti-mouse Cy3; donkey anti-mouse Alexa 647, 1:200; Invitrogen) were incubated in a solution of 1×TBS, 0.1% Triton X-100, 6% FBS serum, and 0.1% BSA for 2 h. To those samples containing fibroblasts, phalloidin TRITC (1:40, A12381; Invitrogen) was added to the secondary antibody solution. After the incubation, the samples were washed thrice (20 min each) with TBS 1×. The nuclei were counterstained with a blue fluorescent Hoechst dye (HOE 33342; Sigma). Samples were mounted between two coverslips with mounting media (Fluoromount-G, 0100-01; Southern Biotech). Images were acquired in a confocal laser scanning microscope (SP-2; Leica).
Statistical analysis
Data from the TER are presented as mean±SE from the three independent experiments. Days 4–6 on the polycarbonate membranes and days 6–8 on the biotranswells, corresponding to the time points at which the cultures reached a plateau, were averaged. A two-way analysis of variance was applied to assess differences between the two types of transwells and the cell culture combination. Multiple comparisons between groups were performed by means of Student–Newman–Keuls post hoc method.
Results
Properties of the acellular membranes
Matrix composition
After decellularizing the pig tracheas, the remaining ECM components and nuclear material were investigated by histological techniques (Fig. 2). van Gieson staining revealed the maintenance within the tunica mucosa and submucosa of the integrity of both collagen and elastin fibers (Fig. 2C). Alcian blue, counterstained with hematoxylin to identify the remaining nuclei, showed that glycosaminoglycans (GAGs) were preserved in the matrix and that in most of the membranes cells were successfully eliminated from the tissue (Fig. 2F). The elimination of the cell component was also confirmed by imaging the elastin fibers in the two-photon microscope. Since samples had been stained previously with Hoechst, any remaining nuclei could also be detected simultaneously. In some tracheas, however, some nuclei were still present in the submucosal glands (data not shown). In the studies presented here, membranes containing cell remnants were discarded.
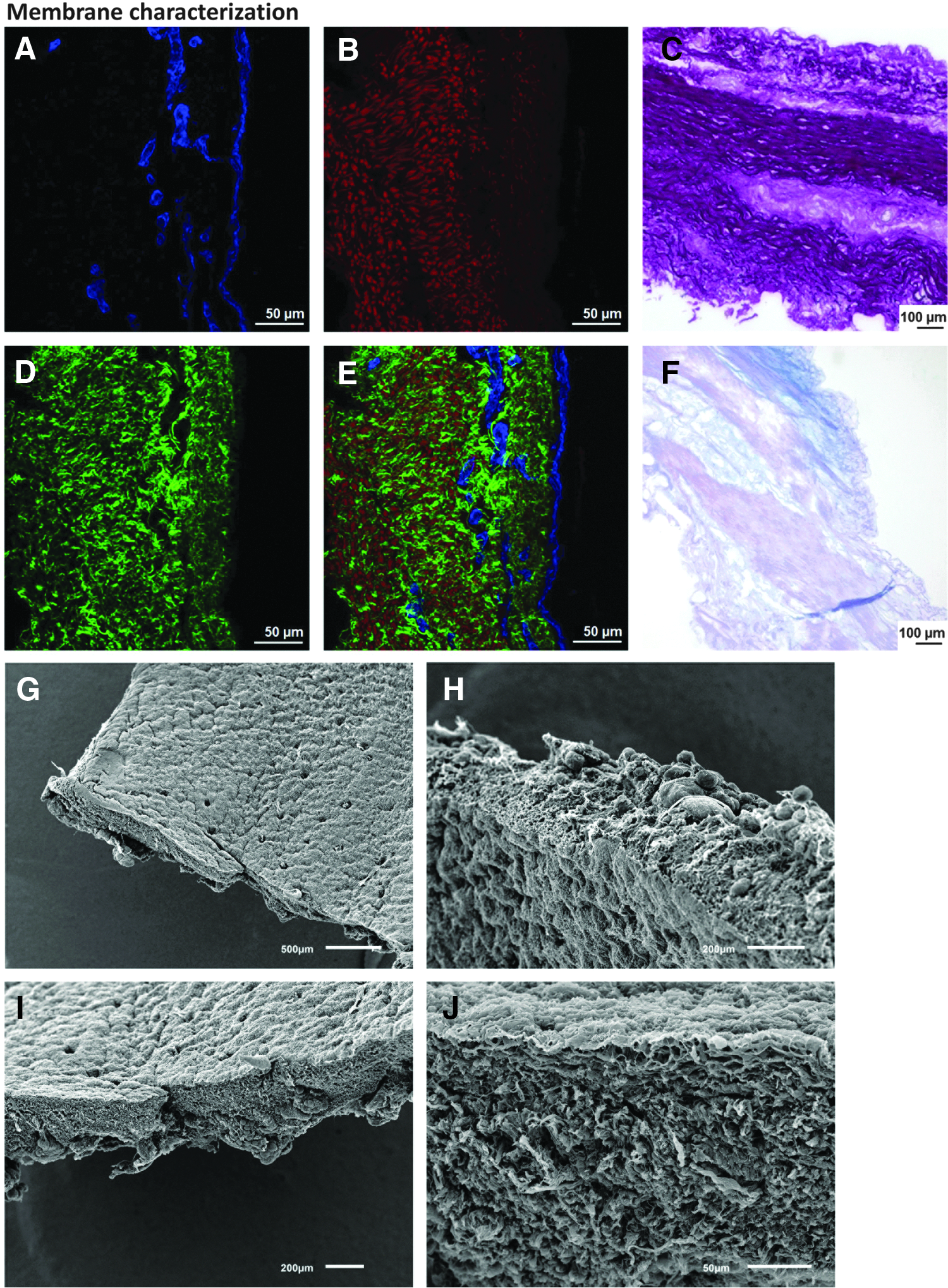
Membrane characterization. Laminin immunostaining
Elastin and collagen fibers were also identified by two-photon microscopy and SHG, revealing the same pattern found using the classical van Gieson staining (Fig. 2B, D). On the same samples, laminin was detected by immunohistochemistry and appeared intact, thus indicating the preservation of the basement membrane (Fig. 2A). Figure 2E depicts an overlay of Figure 2A, B, and D.
To assess further the morphology of the decellularized membranes, SEM was performed on the samples. As expected, the apical side of the membrane appeared smoother than the basolateral side. The apical aspect corresponded to the surface on which the bronchial epithelial cells were growing and where laminin was identified (Fig. 2G, J). The end of the ducts, where the submucosal glands open on to the surface could be observed as small holes in the decellularized membrane (Fig. 2G). The basolateral side was formed mainly by collagen and elastin fibers mixed together (Fig. 2J).
Cell characterization
Cell type markers
Bronchial epithelial cells expressed β-catenin as well as ZO-1 proteins (Figs. 3A–H and 4) on both membrane models (polycarbonate and biotranswell). This indicates that adherent and TJs, respectively, were formed between the cells.

Cell characterization on the biotranswells and polycarbonate transwells. Immunostaining for β-catenin (
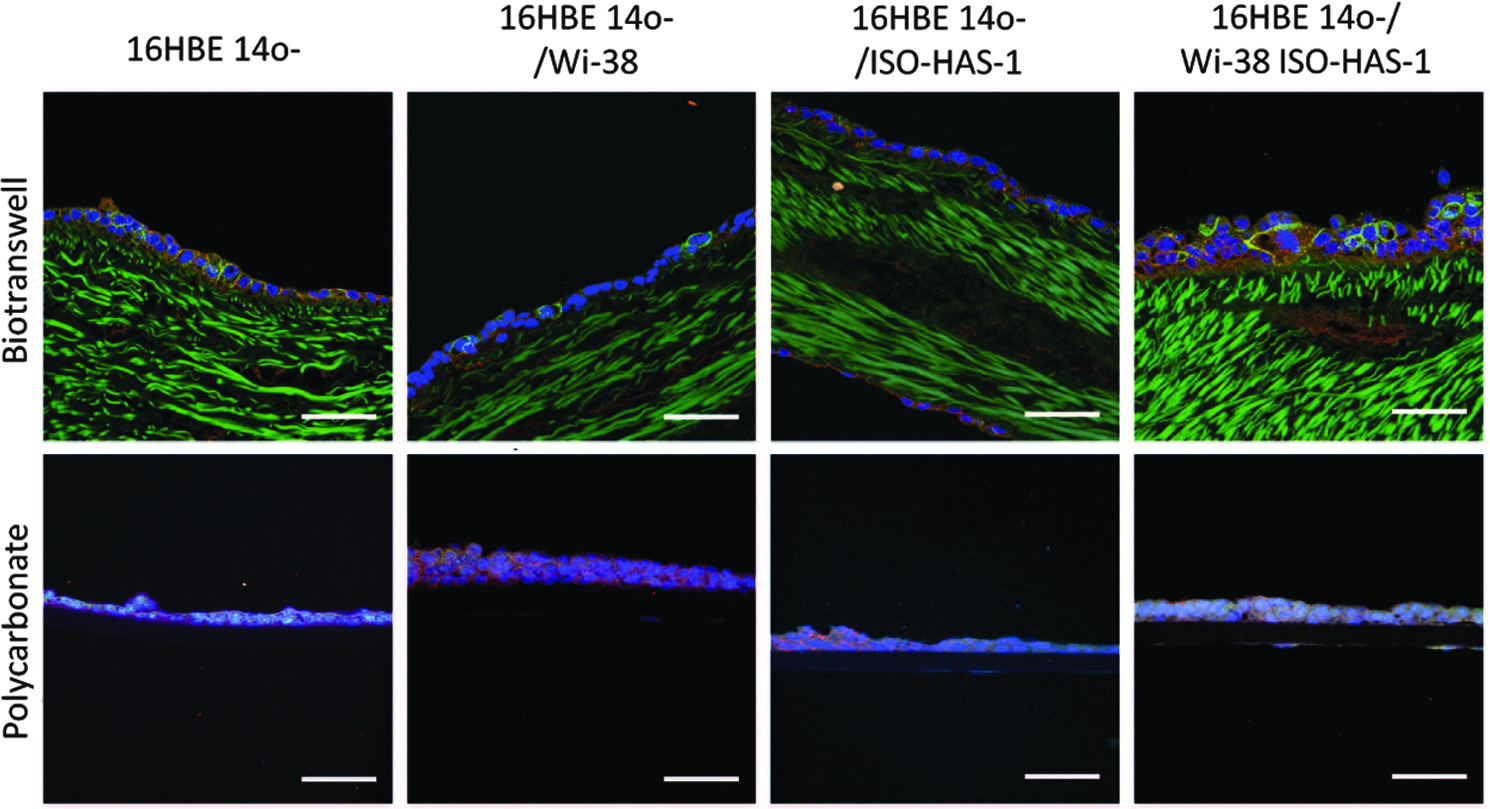
Transverse sections of the transwells. Staining of ZO-1 (green) and β-catenin (red) revealed the formation of a barrier in both cell culture supports. Nuclei are counterstained with Hoechst (blue). Elastin autofluorescence is detected simultaneously when imaging the ZO-1 on the biotranswell samples. Scale bars=50 μm.
On staining the sectioned samples (Fig. 4), we observed that cells grew as a multilayer, being, in most cases, thicker when cultured on the biotranswells. Elastin fibers were visualized simultaneously during imaging of the ZO-1 on account of the intrinsic autofluorescent properties of this structure.
Endothelial cells (ISO-HAS1) detected by the specific expression of CD31, grew on the basolateral side of the polycarbonate membranes and biotranswells (Fig. 3J). They were also present on the cocultures with fibroblasts, Wi-38 (or triple culture), of both transwell types (Fig. 3K).
Fibroblasts were positive for vimentin and had prominent actin filaments stained with phalloidin. They were found growing on the basolateral side of both the natural and synthetic substrates. Since endothelial cells were also positive for vimentin, in the triple culture, fibroblasts were identified by the absence of CD31 staining, but the presence of actin filaments (Fig. 3K).
Scanning electron microscopy
Examination of the samples through SEM revealed the presence of a confluent layer of 16HBEo- on both surface types (Fig. 5). It was observed that the cell density on top of the biotranswells was higher than that on the polycarbonate membrane. Cells growing on the latter appeared larger in surface area and more flattened, although no morphometry was performed to quantify this difference (Fig. 5, images from the apical side). Nevertheless, this was a consistent observation.
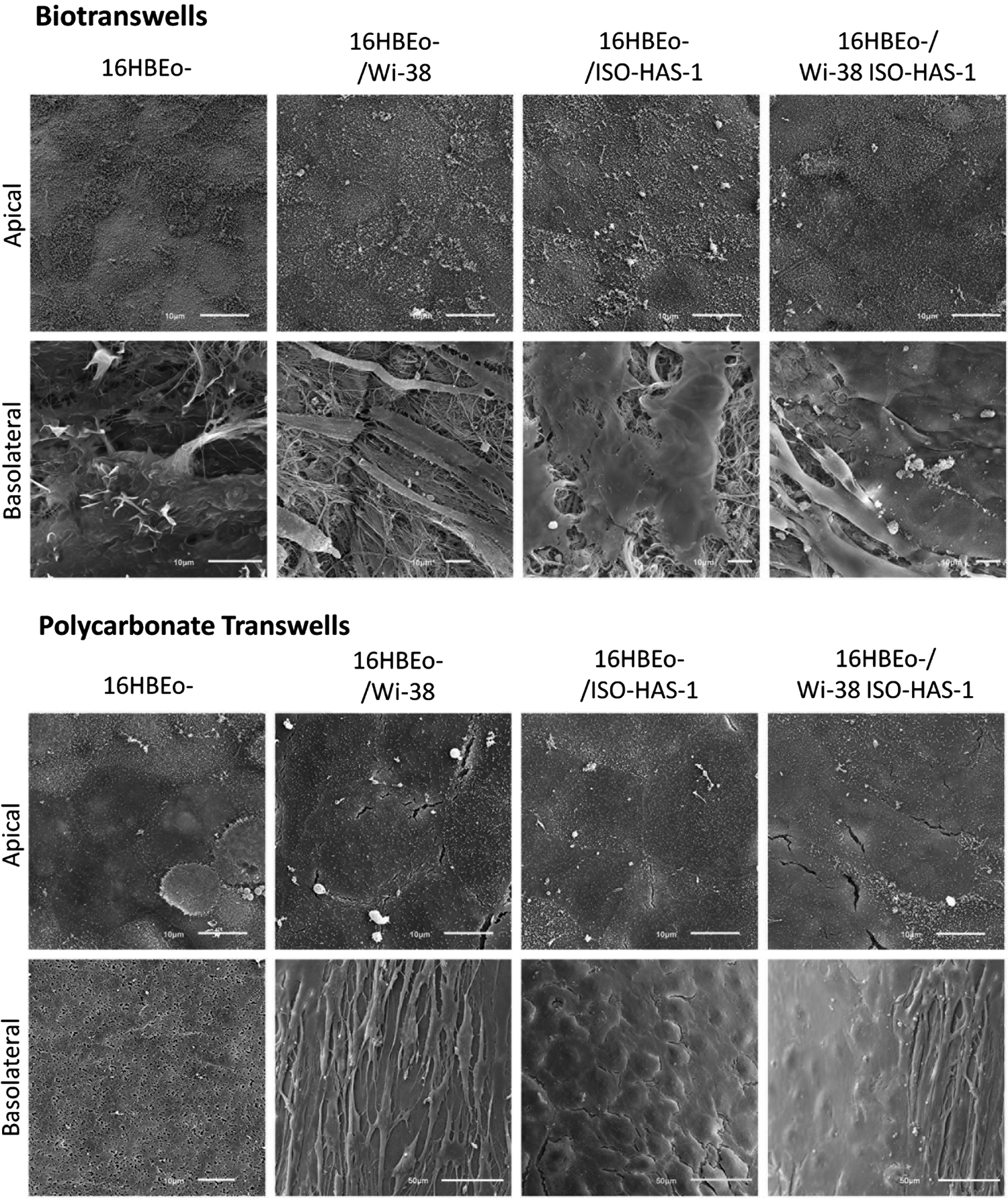
Cell surfaces of the apical and basolateral monocultures and cocultures by scanning electron microscopy. Bronchial epithelial cells appeared larger in surface area and with a flattened shape when growing on the polycarbonate membranes. Biotranswells were more confluent and had an extensive layer of abundant microvilli. Fibroblasts, endothelial cells, and the combination of both cells could be identified on the basolateral side of both transwell types. Scale bars=10 μm.
In addition to this, a remarkable difference between the cells growing on the two different substrates was also detected with respect to the amount and density of the microvilli (Fig. 6A, B). Cells on top of the biotranswell had a dense and homogeneous population of microvilli, whereas those growing on the synthetic material had much less density and appeared shorter. Only a few cells growing on the top of the polycarbonate transwells exhibited a surface pattern similar to those found on cells growing on the biotranswells. Moreover, we observed clusters of cilia on cells growing on the decellularized membrane, indicating that the cell cultures had begun to differentiate toward more mature cells normally found in vivo. (Fig. 6D). Cell–cell contacts were also more evident and defined on the epithelial cells growing in the biotranswell and were seen as distinct paracellular lines (Fig. 6C).
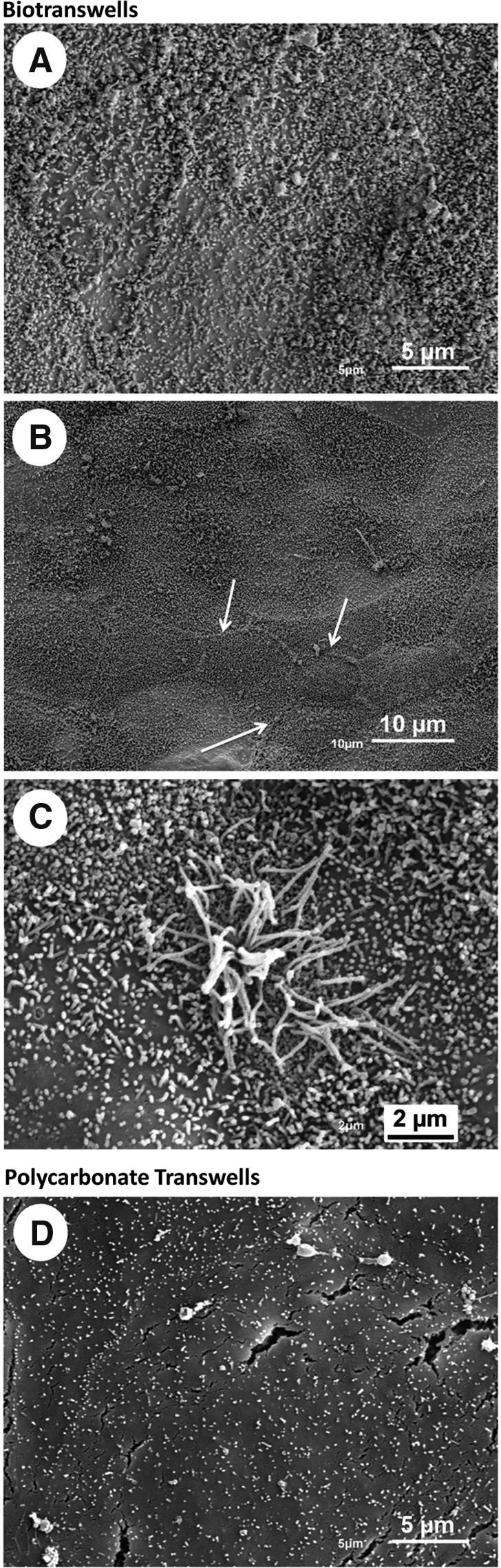
Cells growing on the biotranswells express more microvilli, marked cell–cell contacts, and clusters of cilia in different stages of development. The surface of 16HBE14o- cells growing on the biotranswell was covered with a layer of abundant and much longer microvilli
Endothelial cells (ISO-HAS-1) growing in the respective cocultures and in both types of transwells could be also identified by SEM (Fig. 5, basolateral rows). Fibroblasts (Wi-38) grew on top of the polycarbonate membranes forming a monolayer and in groups along the collagen fibers of the acellular membrane. Fibroblasts were also found growing in coculture with the endothelial cells, indicating that both cell types could coexist as can be observed in vivo.
Transepithelial electrical resistance
TER of the cultures increased over time, reaching a plateau around days 4–6 on the polycarbonate membranes and days 6–10 in the biotranswells (Fig. 7). Subsequently, the TER decreased, especially on the polycarbonate membrane cultures. An average value of the TER at the plateau phase was determined for both membrane models (Fig. 8). On the polycarbonate membranes, 16HBE14o- in monoculture reached 356 Ω·cm2 (±29, SEM). The TER increased substantially for the coculture with fibroblasts, Wi-38 (518 Ω·cm2±49, SEM), and even more for the coculture with endothelial cells, ISO-HAS-1 (648 Ω·cm2±56, SEM). In the triple culture of 16HBE14o- with fibroblasts and endothelial cells, a TER of 665.5 Ω·cm2 (±59, SEM) was detected. The monoculture of 16HBE14o on the biotranswell reached higher values than those on the synthetic transwell for the same condition: 555.24 Ω·cm2 (±31, SEM). In addition, in the biotranswell, cocultures with fibroblasts, Wi-38, reached a TER value of 565 Ω·cm2 (±41, SEM) and with endothelial cells, ISO-HAS-1, this reached 638 Ω·cm2 (±37, SEM). The triple culture achieved a TER of 552 Ω·cm2 (±38, SEM). However, there was no significant difference between the two transwell types for co- and triple cultures (p=0.319). A difference between the transwell types was established only for the 16HBE14o monoculture, as described above (p<0.05). As for the differences between the coculture conditions, these were significant when comparing monocultures with cocultures (p<0.05). ISO-HAS-1 and Wi-38 alone did not exhibit any TER (data not shown).
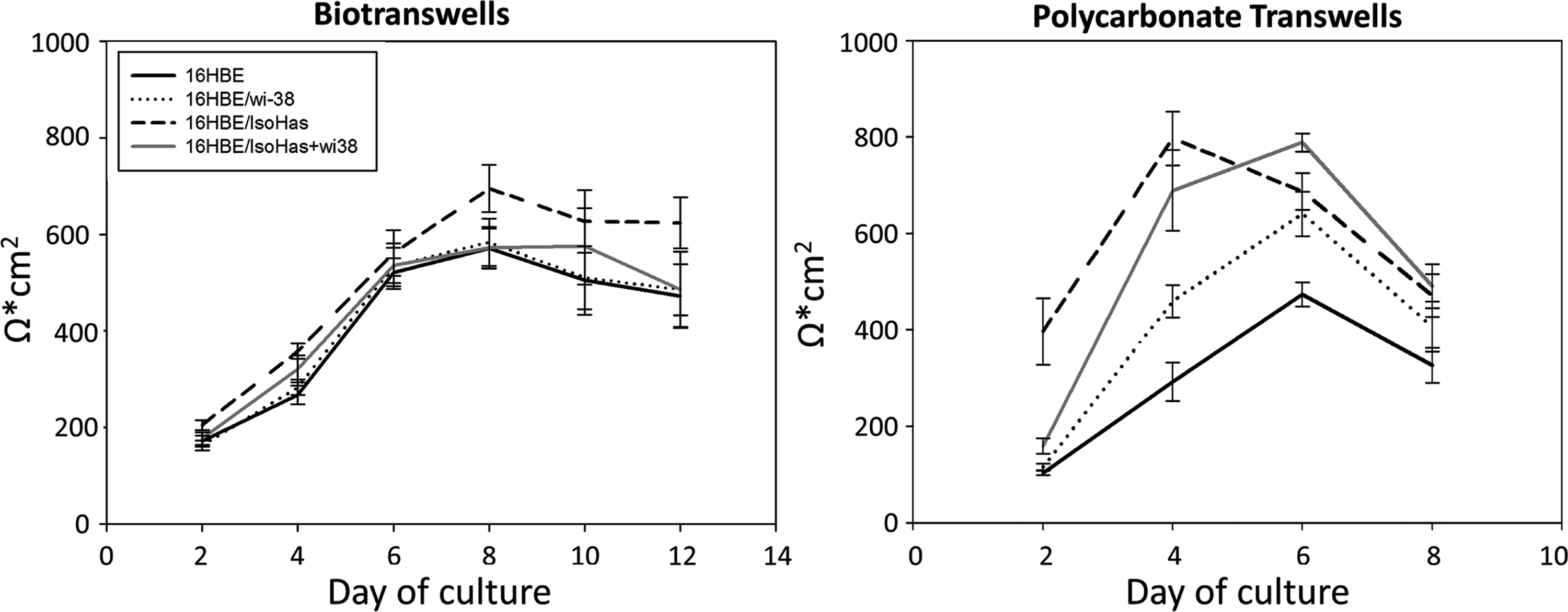
Time course of the development of transepithelial electrical resistance (TER). Monocultures of 16HBE14o- cells grew to form a barrier in both transwell inserts, reaching the highest values on days 6–8 on the biotranswells and days 4–6 on the polycarbonate transwells. Coculture with fibroblasts (Wi-38) and/or endothelial cells (ISO-HAS-1) increased the values compared with those observed in the monoculture. Data are pooled from three independent experiments.
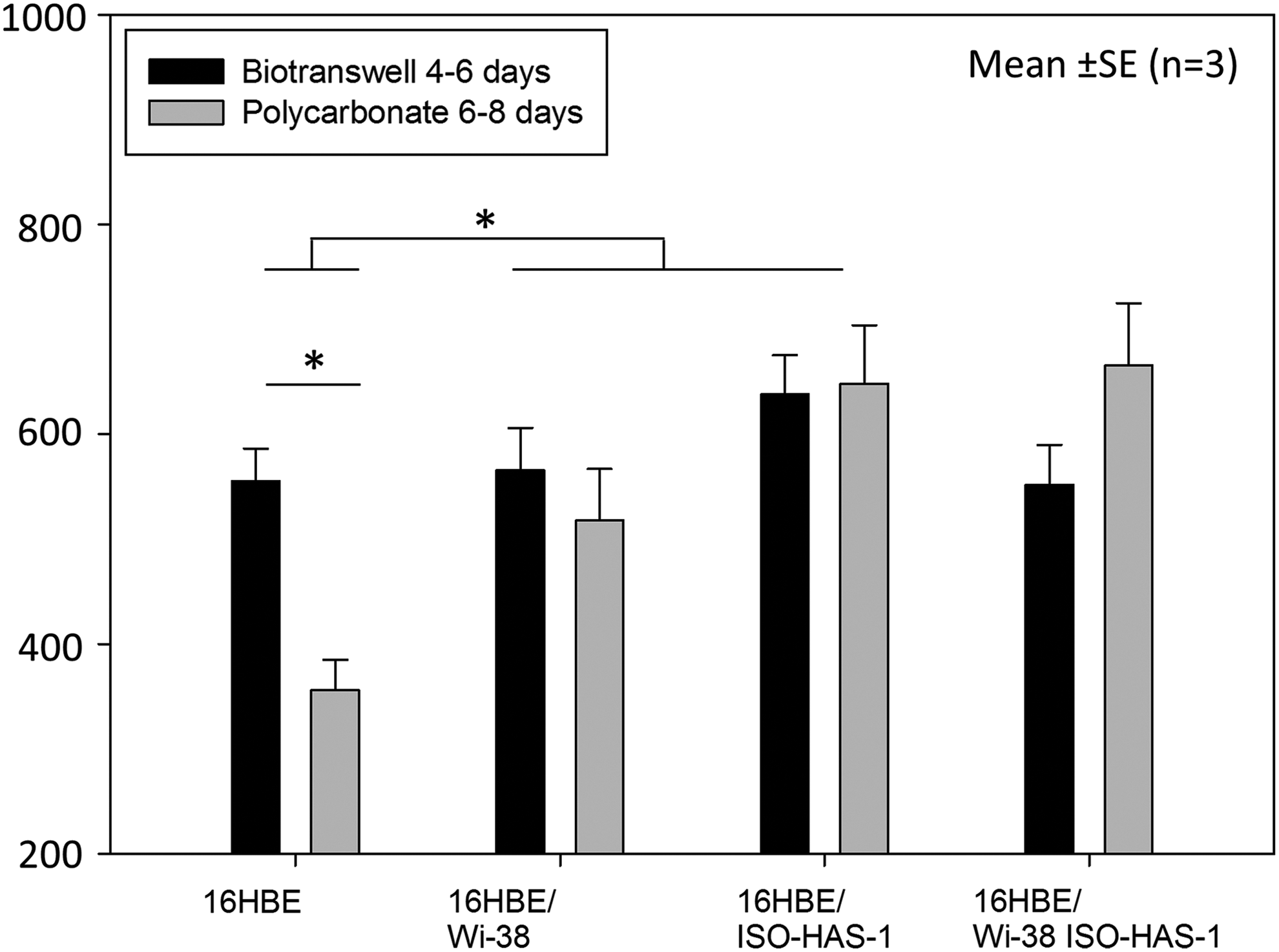
TER value averages on the plateau days. Mean values of the different cultures for days 4–6 (polycarbonate) and days 6–8 for the biotranswell. There is a significant difference between the supports when cells are grown in monoculture (p<0.05). Significant differences can be observed when comparing monocultures and cocultures with fibroblasts (Wi-38) and endothelial cells (ISO-HAS-1) p<0.05. Each data point represents the mean±SE of three independent experiments. For statistical analysis two-way ANOVA with Student-Newman Keuls post-hoc test was conducted. *P<0.05.
Discussion
The results from this study demonstrated that decellularized tracheal tissue can be successfully employed for multicellular bronchial in vitro models. Using three relevant cell types in coculture, such as epithelial cells, endothelial cells, and fibroblasts, in combination with such a native biomembrane, mimics more closely the in vivo situation.
After the decellularization process, the resulting tissue was a highly elastic membrane. It permitted easy handling and stability during the various manipulation steps, such as cell seeding and culturing, as well as various subsequent processing steps concerning analysis and characterization of the recellularized in vitro construct.
According to van Gieson staining and SEM, a thin matrix of mixed collagen and elastin fibers is located beneath the basement membrane of the bronchial epithelium. Additionally, laminin is preserved and colocated with the basement membrane of the native bronchial epithelial cells. This indicates that this crucial in vivo structure, involved in the attachment, motility, differentiation, and polarization of the epithelial cells,37–41 is maintained after the decellularization steps. Laminin remained as a thin layer on top of the collagen and elastin fibers and represents the component that 16HBE14o- cells encounter first when seeded on top of the transwells. On the other hand, elastin and collagen fibers, in addition to other proteins, were the substrate for the endothelial and/or fibroblastic cells growing on the lower side of the biomembrane (directed to the basolateral compartment). Thus, this biotranswell model was formed with a tissue-based membrane that contains the protein components of the ECM distributed in the same manner as occurs in the bronchial microenvironment in vivo. 42
It was observed that GAGs were maintained within the matrix. Although in most of the samples the decellularization was accomplished successfully, in certain tracheas cellular remnants were found in the mucosal glands. These glands appear to display a higher barrier for the CHAPS to penetrate the glandular tissue to remove the cells. These results have also been reported by other authors. 43 For the studies reported here, samples were stained with Hoechst and those that were positive for DNA were discarded so that only fully decellularized tissues were employed for the construction of the biotranswells. To gain more insight into the suitability of the native construct for multicellular bronchial in vitro models, we compared them to the synthetic transwell systems, which are based on a semipermeable polycarbonate filter membrane, this being the usual model for such experimental purposes.12,13
After recellularization of the matrix membrane by 16HBE14o, ZO-1 and β-catenin were well developed in the confluent epithelial layers on both the synthetic and natural substrates. This indicates an optimal differentiation and polarization of the epithelium.44,45 These junctions were also visible by SEM and were very prominent in the case of the cultures growing on the biotranswell, as is shown in Figure 6C. Protruding paracellular lines have also been reported for other lung epithelial cells and correlated with a polarized architecture. 11 As observed by IF (Actin, vimentin, CD31) and SEM, fibroblasts (Wi-38) and endothelial cells (ISO-HAS-1) grew well in coculture on the basolateral side of the biomembrane and in triple culture having 16HBE14o on the apical side. Fibroblasts were additionally stained for smooth muscle actin, giving a negative profile (data not shown). This would indicate that these cells were not activated fibroblasts and that contractile activity was not increased. 46
Fibroblasts growing on the biotranswell had a more in vivo-like appearance (more robust bodies and less thin projections) than those on the polycarbonate membrane. One possible explanation for this could be the fact that on the polycarbonate transwell, cells are growing on a collagen-treated synthetic surface, which is foreign in composition to what cells encounter in vivo. Several crucial proteins are lacking that are known to alter the behavior of the fibroblasts. 42 Another explanation could be that the surface of the biotranswell on the basolateral side matches more closely the mechanical properties needed by the fibroblasts. It has been demonstrated that sites in the lung, which are enriched in collagen, and host fibroblast-like cells (submesothelial cells and pericytes) appear to have similar micromechanical behavior when measured with atomic force microscopy. 47
Endothelial cells were able to grow on both substrates. However, labeling of CD31 revealed a more regular morphology of the cells on the polycarbonate compared with the cells on the biotranswell. This could probably be due to the fact that the surface of the polycarbonate membrane is smoother and more homogeneous compared with the biotranswell. More detailed studies on endothelial functionality are required to compare the endothelial phenotype on both substrates.
To assess possible differences between the culture conditions, TER was monitored over a number of days. An increase in TER was correlated with the development of the junctions between the cells.17,18 We found that cells growing on both transwell types reached values similar to those described in the literature for 16HBE14o- in submersed cultures. 48 It was also observed that cells growing on the polycarbonate samples reached a plateau much faster (4–6 days depending on the culture) than cells growing on the decellularized membranes (6–8 days). This accelerated process could be attributed to the higher proliferation of human airway epithelial cells when growing on collagen-coated surfaces. 42 Alternatively, if cells are grown in Matrigel®, a preparation that contains the main components of the basement membrane (laminin 61%, collagen IV 30%, and entactin 7%),49,50 cell proliferation is found to be reduced. 42 Therefore, it is reasonable that cells proliferate slower in the biotranswell since it provides the natural basement membrane, resulting in a longer period of time to reach a confluent state. However, a higher proliferation rate does not necessarily indicate a better viability or functionality of the cells since we are dealing with permanent transformed cell lines derived from human tumors, which usually exhibit a high proliferative rate. With respect to recellularization of tracheal grafts in the future, it should be stressed that the aim is to have a controlled proliferation while achieving optimal cellular differentiation, polarization, and functionality.
Although cultures reached a plateau of TER much faster in the case of the synthetic membranes, the bronchial epithelial cell line retained high values on the biotranswell over a longer period of time (6 days). The TER decayed slightly after day 8, but not as quickly as on the polycarbonate transwells. One possible explanation for this observation could be the effect of the substrate composition. In this case, the preserved basement membrane of the decellularized membrane contributed to a higher differentiation and polarization of the epithelial cells, which result in a tighter barrier and tight cell–cell contacts. 51 It is probable that the decrease of TER in the conventional transwell models with epithelial cell lines on a polycarbonate membrane is a result of the lost ability of contact inhibition and the continuing proliferative activity.
Collagen matrices have often been used as a simplified in vitro basement membrane in airway epithelial cell culture models, but neglect the fact that epithelial cells in vivo grow on a more sophisticated and heterogeneous basement membrane than on type I collagen alone. Many studies have highlighted the importance of the basement membrane with respect to the maintenance of airway epithelial integrity. This is mirrored in several obstructive lung diseases such as asthma, in which its composition is altered.42,52
Another example is a study on rat basal cells, which when cultured on a collagen-I-coated substrate kept a squamous nonpolarized phenotype, whereas on a synthesized basement membrane substratum, they successfully polarized and differentiated into ciliated cells. 53
In addition, in the present study, a better differentiation and degree of polarization of the bronchial epithelial cells cultured on the biomembrane were corroborated by SEM. At a higher magnification, well-developed microvilli become obvious on 16HBE14o growing on biotranswells compared with the cells on the polycarbonate membrane (Fig. 8A, B). Moreover, clusters of emerging cilia were found on the cells growing on the biomembrane, which is a further indicator of a more differentiated state. 48
Based on this, the different behavior of the cells in terms of TER pattern and the state of polarization/differentiation found in our studies could be attributed to the favorable and in vivo-like composition of the decellularized substrate. Taking these results together, it can be concluded that the biotranswell is more appropriate than the polycarbonate membrane for the growth and differentiation of respiratory epithelial cells. When averaging the data of the days corresponding to the highest TER value in both systems (days 4–6 for the polycarbonate transwells and days 6–8 for the biotranswells), we established a significant difference between the monocultures growing on the biotranswell with a value of 555.24 Ω·cm2 (±31, SEM) compared with the synthetic membrane with 356 Ω·cm2 (±29, SEM) (p<0.05). This could explain why the effect of the cocultures is more obvious on the polycarbonate membranes as the monocultures on this substrate are growing in a more unsuitable environment that could be improved by the addition of another cell type. In the case of the monocultures growing on the biotranswell, the effect of the coculture would not be so evident due to the fact that the native basement membrane itself would enhance the barrier properties of 16HBE14o- significantly.
Nevertheless, taking the data from both substrates together, a clear difference between the monocultures and the cocultures can be seen (p<0.05), with the TER values in the cocultures being substantially higher than those measured in the monocultures. This result reinforces the hypothesis that cells in coculture have a paracrine interaction and that the release of mediators by endothelial cells or fibroblasts enhances the properties of the epithelial barrier.12,13 The nature of these products of cellular cross talk still needs to be elucidated.
Bronchial tumor epithelial cells or immortalized primary cells, such as 16HBE14o- cells or BEAS-2B, are commonly used in studies of the upper respiratory airway. While exhibiting suitable respiratory bronchial epithelial cell-like phenotypes, 54 these cells have the advantage of being a clonally identical cell type (not a mixed population). Moreover, they can be synchronized, expanded to high numbers (permitting biochemical and screening studies), and can also be used to measure epithelial fluxes and TER. 15 Although the results and observations cannot be readily extrapolated to the native tissue, they could be used to examine the direct effect that media composition, growth factors, or drugs may have on the behavior of normal cells.
In general, primary cell culture models are not routinely used for biopharmaceutical purposes 45 due to the low availability of primary tissue, low yields of isolated cells, and the problem of high biological variability. On the other hand, it is known that normal synthetic transwell models do not sustain differentiated primary cells. After some days in culture, these cells lose their differentiated phenotype, probably due to the lack of ECM, growth factors, and hormones. 55 This problem could be overcome with the use of the biotranswell, in which natural components of the ECM are retained. In this study, we have shown that there is reciprocity between the ECM and the differentiated state of resident cell populations, which would confer a relevant advantage in the use of decellularized materials compared with synthetic materials. 24 These natural materials provide the cells with an appropriate ECM architecture and composition as they would find in their natural environment, which is location specific and whose isoforms vary along the entire respiratory tract. 23
Native membranes also provide an advantage for tissue engineering purposes, and it has been shown that recellularization of decellularized scaffolds can be a successful strategy.6,7 As previously reviewed by Bader and Macchiarini, several attempts have already been made to transplant recellularized tracheal allografts in humans, but face the constant challenge of avoiding ischemic changes. 7 This normally results from an insufficient revascularization, which fails to compensate for the deficient graft blood supply. Thus, the multicellular, bronchial in vitro biotranswell described in this study could be the ideal construct to conduct fundamental research with respect to proper tissue regeneration, including a sufficient revascularization. Steinke and coworkers used a native scaffold, which is a decellularized, porcine small bowel segment. In this construct, the scaffold of the capillary network is well preserved and could be successfully revascularized and recellularized in vitro and implanted in human patients for tracheobronchial reconstruction, as reviewed by Steinke. 56 Prospective studies combining this recellularization technology with our native tracheal scaffolds could therefore be a benefit for successful transplantation of tracheobronchial allografts in human recipients.
Conclusion
In this work, we have successfully generated an experimental, bronchial coculture model based on a natural scaffold, which displays more in vivo-like properties compared with commercial, synthetic transwell models. The feasibility of the biotranswell production makes it convenient for routine research, for the moment using cell lines, but hopefully in a next step with primary cells.
In summary, the biotranswell presented in this article opens up the possibility of assessing multiple parameters that influence the bronchial epithelial barrier (including drugs and toxic agents) as well as the recellularization processes, which are as yet inadequately understood, but crucial to tissue engineering and regenerative medicine approaches.
Footnotes
Acknowledgments
The authors wish to thank Ms. Elke Hübsch and the scientific and technological centers from the University of Barcelona (advanced optical and electronic microscopy) for the excellent technical assistance. This work was supported, in part, by the Spanish Ministry of Economy and Competitiveness (SAF2011-22576) and DFG priority program SPP 1313 within the Cluster BIONEERS (RepairLab) and state funds from Rhineland-Palatinate.
Disclosure Statement
No competing financial interests exist.