
Abstract
Stem cells expressing reporter constructs are extremely useful for their tracking in vivo or for determining cell lineage fate in vivo and in vitro. We generated liver progenitor cell (LPC) lines from actin-EGFP and TAT-GRE-lacZ mice. LPCs from the actin-EGFP mouse facilitate cell tracing following transplant as the reporter is constitutively expressed. LPCs from the TAT-GRE-lacZ mouse express β-galactosidase under the control of the tyrosine aminotransferase (TAT) promoter and are only active in mature hepatocytes. We found that the utility of such LPC lines becomes severely limited by downregulation of transgene expression following extended culture. We show that epigenetic mechanisms are responsible for suppressing expression of both transgenes. Enhancement of transgene expression in both LPC lines was achieved by treating the cell lines with either the histone acetylating agent sodium butyrate or the DNA demethylating agent 5-azacytidine.
Introduction
T
NaB and 5-aza C have been used to investigate the direct and indirect effects of induced epigenetic changes in chromatin acetylation and DNA methylation, respectively, in many cell types, including both normal and malignant cells.4–6 NaB has been reported to inhibit proliferation, stimulate differentiation, and restore gene expression in cultured cells.7,8 Treatment of cells by NaB results in histone hyperacetylation by inhibiting histone deacetylases.9,10 This induces histone H3 acetylation and promotes DNA demethylation, which alters expression of pluripotency-associated genes. 11 In contrast, 5-aza C stimulates the removal of methyl groups from DNA by inhibiting DNA methyltransferases,12,13 which can induce gene expression, including genes that regulate differentiation.14,15
Liver progenitor cells (LPCs) are considered to be facultative hepatic stem cells that can differentiate into functional hepatocytes and cholangiocytes in the setting of chronic liver injury.16,17 LPCs have assumed increased significance given their therapeutic potential, that is, their stem-cell like ability to regenerate the liver.18,19 In contrast, the application of mature hepatocytes for therapeutic intervention is hampered by their inability to proliferate and maintain function when expanded by long-term in vitro culture. 20
To study hepatocyte differentiation from LPCs, cell lines were generated from the TAT-GRE-lacZ mouse and designated BMEL-TAT. 21 BMEL-TAT expresses β-galactosidase under the control of the liver-specific tyrosine aminotransferase (TAT) promoter, which is only expressed in mature hepatocytes. 22 In this multicopy transgene model, the β-galactosidase reporter faithfully reflects the endogenous expression of liver-specific TAT. 23 Unexpectedly, the utility of this cell line was compromised when transgene expression was diminished after they were extensively expanded in long-term culture.
To ascertain whether epigenetic mechanisms were responsible, we assessed the effects of the NaB and 5-aza C on the activity of the β-galactosidase reporter gene in BMEL-TAT cells. We show that both agents were able to enhance reporter gene expression. However, in this model, we were unable to ascertain whether epigenetic modulation suppressed the ability of the cells to differentiate or whether they have downregulated expression of the reporter. To test whether a reporter gene that is expressed independently of the differentiated status of the LPC is attenuated by epigenetic mechanisms, we generated multicopy transgenetic LPCs from the actin-EGFP mouse 24 where constitutive EGFP expression is regulated by the actin promoter and designated as BMEL A-EGFP. We show that NaB and 5-aza C enhance EGFP reporter expression in these LPCs. We also show that NaB and 5-aza C reactivate reporter expression in extensively passaged (p57) BMEL A-EGFP cells, in which the transgene was almost completely extinguished. We conclude that epigenetic mechanisms operate to downregulate reporter genes in progenitor/stem cells as they are passaged and this reduces their utility. Epigenetic modification can increase the activity of the transgenes and restore their utility.
Materials and Methods
LPC isolation
Livers from 14-day gestation mice were washed with a warm (37°C) balanced salt solution (0.5 mL per liver; BSS), and lobes were separated and freed from the adjoining gut and mesenchyme using fine forceps. Lobes from each liver were incubated in 0.5 mL of BSS containing 0.6 mg of collagenase H (Boehringer Mannheim) for 20 min at 37°C with gentle mixing every 5 min. Following centrifugation for 5 min at 300 g, the collagenase solution was replaced with a growth medium that comprised Williams' E medium (Sigma-Aldrich) supplemented with 2.5 μg/mL Fungizone (Life Technologies), 48.4/675 μg/mL penicillin/streptomycin (Sigma-Aldrich), 2 mM
Establishment and culture of LPC lines from primary cultures
LPC lines were established using the protocol of Strick-Marchand and Weiss. 25 Primary cultures were maintained in the growth medium. During extended culture (8–12 weeks), most cells senesced and occasional epithelioid colonies emerge. These were selected and subcultured using cloning rings. By this approach, the BMEL-TAT and BMEL A-EGFP LPC lines were derived from TAT-GRE-lacZ and actin-EGFP transgenic mice, respectively. Once established, these LPC lines were passaged routinely when they attain 70–80% confluency in a growth medium containing 5% FBS and they did not require collagen-coated culture dishes to attach and grow.
LPC differentiation protocol
Aliquots of 3×105 LPCs were transferred to 35-mm dishes, made up to 2 mL with growth medium, and maintained for 10 days to reach 100% confluency, with medium replacement every 2 days. The growth medium was then substituted with a differentiation medium (Williams' E containing 2.5 μg/mL Fungizone, 48.4/675 μg/mL penicillin/streptomycin, 2 mM
Treatment of LPCs with NaB and 5-aza C
Cells were treated with 5 mM NaB (Sigma-Aldrich) or 4 μM 5-aza C (Sigma-Aldrich) by addition of a 500 mM or 400 μM respective stock solution to the culture medium. The stock solutions were prepared in a 0.1% dimethyl sulfoxide (DMSO) vehicle (Sigma-Aldrich). Cultures were treated daily for 3 days.
Immunofluorescence staining of LPCs
Anti-EpCAM (ab71916; abcam), anti-E-cadherin (3195; Cell Signaling Technology), anti-vimentin (MAB2105; R&D Systems), and anti-HNF4α (Hepatocyte nuclear factor 4 alpha, sc-6556; Santa Cruz) antibodies were used to stain cells grown to at least 70% confluence on collagen-coated glass coverslips. Cells were fixed in cold (−20°C) acetone–methanol (1:1) for 2 min. After three washes with phosphate-buffered saline (PBS), coverslips were blocked with 1% bovine serum albumin (BSA; Sigma-Aldrich) in PBS for 30 min and then incubated with the corresponding primary antibodies (at a 1/200 dilution in 1% BSA in PBS) overnight (16 h) at 4°C. Coverslips were then washed thrice with PBS followed by incubation with the appropriate secondary antibodies (diluted 1/200 in 1% BSA in PBS) for 1 h at room temperature. Anti-E-cadherin, anti-EpCAM, and anti-HNF4α primary antibodies were labeled with the Alexa Fluor 488 goat anti-rabbit IgG (H+L) secondary antibody (Invitrogen) and anti-Vimentin with the Alexa Fluor 594 goat anti-mouse IgG (H+L) secondary antibody (Invitrogen). Nuclei were stained with 25 ng/mL 2-(4-amidinophenyl)-1H -indole-6-carboxamidine (DAPI; Sigma-Aldrich) in PBS for 5 min at room temperature. After three washes with PBS, coverslips were mounted with the Gelvatol mounting medium (0.2 M Tris [pH 8.5], 30% glycerol, 1% sodium azide, 105 mg/mL poly vinyl alcohol) and viewed with an Olympus IX2-ILL100 Inverted Microscope (Olympus Corporation).
X-gal staining
To assess differentiation based on the β-galactosidase reporter, X-gal (5-bromo-4-chloro-3-indolyl-D-galactopyranoside) staining was performed to determine the percentage of LPCs that differentiated into hepatocytes. Cells were grown to 100% confluence, washed in prewarmed (37°C) BSS, and fixed in 0.25% glutaraldehyde, 0.1 M phosphate buffer (pH 7.3), 5 mM ethylenebis (oxyethylenenitrilo) tetraacetic acid (EGTA), and 2 mM MgCl2 for 3 min at room temperature. Cells were subsequently washed with a solution of 0.1 M phosphate buffer (pH 7.3), 0.01% sodium dodecyl sulfate (SDS), 0.02% Nonidet-P40 (Sigma-Aldrich), and 2 mM MgCl2 for 3 min at room temperature, followed by three washes in PBS. To visualize β-galactosidase, cells were incubated overnight in X-gal staining solution (0.1 M phosphate buffer [pH 7.3], 20 mM Tris buffer [pH 7.3], 0.01% SDS, 0.02% Nonidet-P40, 2 mM MgCl2, 5 mM K3[Fe(CN6)], and 1 mg/mL X-gal) at 37°C. The staining was visualized and recorded using a Nikon Eclipse TS100 inverted microscope (Nikon Corporation).
Fluorescent β-galactosidase assay
Cells were collected by centrifugation (1000 g for 5 min), washed with BSS, and resuspended in a cell suspension buffer (40 mM Na2HPO4, 60 mM NaHPO4, 10 mM KCl, and 1 mM MgSO4). Extracts were collected by centrifugation at 15,000 g for 10 min at 4°C and aliquots were taken for enzymatic and protein measurements. Protein concentration was determined using the Bio-Rad Bradford Protein Assay kit (Bio-Rad) by mixing 2 μL of extract with 798 μL of diluted reaction mix and 200 μL of reagent in plastic cuvettes. Absorbance was measured in the Eppendorf BioPhotometer at 600 nm along with BSA protein standards.
β-galactosidase assays were conducted in microtiter plates and used 200 μL reaction volumes, each containing 2 μL of cellular extract and 198 μL of 4-methylumbelliferyl-β-D-glucuronide (MUG) substrate solution (51 mg of MUG dissolved in 10 mL of cell suspension buffer and 90 mL of sterile water). Measurements were taken every 2 min for a total of 2 h, and the β-galactosidase activity was determined by averaging the rate of 4-methylumbelliferone (MU) production. All experiments were repeated at least thrice. Fluorescence was determined using the FLUOstar OPTIMA plate reader (BMG Labtech GmbH; filter set: 370 nm excitation and 460 nm emission).
ImageScope quantification
To determine the average X-gal staining and EGFP intensity, fluorescent images were collected from 10 random cells within hepatospheres or 10 fields of view, respectively, for each cell line or treatment group using the Olympus IX2-ILL100 Inverted Microscope (Olympus Corporation) and analyzed using the Positive Pixel Count Algorithm with the Aperio ImageScope software (Aperio Technologies). For EGFP intensity, this process was repeated every 24 h for 3 days to assess the effect of each treatment with time.
Flow cytometry
Cells were washed with BSS, treated with trypsin–ethylenediaminetetraacetic acid (EDTA; Invitrogen), and then resuspended in PBS. Samples were analyzed using the FACSCalibur Flow Cytometer (Becton Dickinson) and CellQuest Pro software (Becton Dickinson). Dead cells were excluded from analysis by forward- and side-scatter gating. A minimum of 9000 events were collected for each sample.
Results
LPCs express epithelial and not mesenchymal markers
The epithelial phenotype of the LPCs was confirmed by expression of E-cadherin and EpCAM surface markers. BMEL-TAT and BMEL A-EGFP LPCs express E-cadherin (Fig. 1, a and j) as well as EpCAM (Fig. 1, d and m). BMEL-TAT is negative for the mesenchymal cell surface marker vimentin (Fig. 1, g and i) and the majority (>95%) of BMEL A-EGFP cells were vimentin negative (Fig. 1p), with rare cells showing positivity (data not shown).

Low-passage BMEL-TAT LPCs (p9) and BMEL A-EGFP (p10) express epithelial and not mesenchymal cell markers. BMEL-TAT
Differentiated BMEL-TAT LPCs express HNF4α and the β-galactosidase reporter, but lose the ability to express this reporter after extended passaging
To demonstrate the utility of the TAT-driven β-galactosidase reporter, passage (p)9 BMEL-TAT LPCs were differentiated and stained with X-gal. Differentiated cells were larger (Fig. 2a, arrowed), with the larger cells having substantially more β-galactosidase activity as indicated by the blue staining (Fig. 2a). Differentiated cells formed spheroid clusters that we designate as hepatospheres; all cells within the hepatospheres stained positively with the majority of cells staining intensely (Fig. 2b). Undifferentiated BMEL-TAT LPCs did not stain (results not shown) and are similar to the negative cells that surround the hepatosphere (Fig. 2b). Differentiated p26 cells show significantly fewer positive cells both in monolayers (Fig. 2c) and in hepatospheres (Fig. 2d). Furthermore, the average size of hepatospheres in low-passage LPCs was 2.4-fold higher than high-passage hepatospheres (Fig. 2B). The differentiated status of the BMEL-TAT LPCs was confirmed by expression of the mature hepatocyte marker, HNF4α. BMEL-TAT cells maintained in the growth medium did not express HNF4α (Fig. 2, e–g), while differentiated LPCs expressed nuclear HNFα (Fig. 2, h–j).
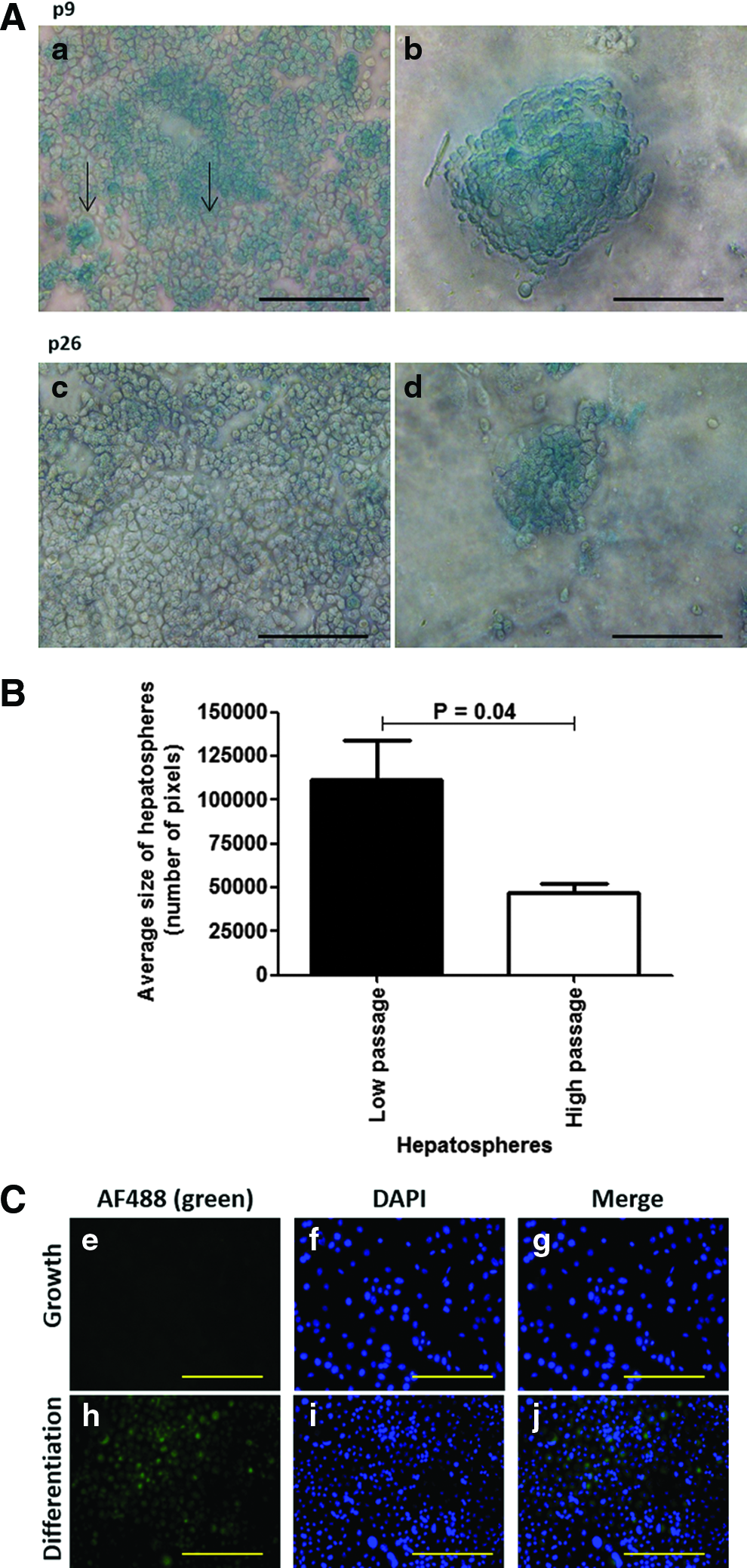
The β-galactosidase activity was measured to quantify reporter expression in differentiated BMEL-TAT LPCs at p9, p16, and p26. A decrease of reporter expression with increasing passage number (Fig. 3A) was evident. The level of β-galactosidase reporter expression was 5.19, 4.08, and 3.30 mU/mg protein for BMEL-TAL LPCs p9, p16, and p26 cells, respectively.

β-galactosidase activity in differentiated BMEL-TAT LPCs declined with extended culture.
The level of β-galactosidase activity in undifferentiated BMEL-TAT LPCs was very low (0.51 mU/mg protein, data not shown). This is similar to the activity of BMEL A-EGFP LPCs (0.24 mU/mg protein), which do not have this reporter construct. Following differentiation, the activity was 4.73 mU/mg protein representing a substantial increase of nearly 10-fold relative to the negative control.
To determine whether the relative X-gal staining intensity of hepatosphere cells changed with increasing passage, BMEL-TAT LPCs were analyzed by ImageScope analysis software. High-passage (p26) cells within hepatospheres showed a decrease in average pixel intensity of 15.4% compared to low-passage (p9) cells within hepatospheres (126±0.9 to 145.4±0.9; Fig. 3B). Furthermore, phase-contrast microscopy shows low-passage BMEL-TAT hepatospheres (Fig. 3C) to be visibly smaller than the high-passage equivalents (Fig. 3D).
Treatment with NaB and 5-aza C increases β-galactosidase activity in differentiated BMEL-TAT LPCs
X-gal staining was more intense in differentiated LPCs that were treated with either NaB (Fig. 4b) or 5-aza C (Fig. 4c) compared with the DMSO-treated controls (Fig. 4a). Notably, a subset of NaB-treated LPCs displayed higher levels of X-gal staining than the majority of the culture (Fig. 4b). In contrast, 5-aza C-treated LPCs have fewer intensely staining cells, but instead showed many more cells with a high level of staining throughout the culture (Fig. 4c). The total number of positive cells was not significantly different in the respective cultures (data not shown); NaB and 5-aza C increased β-galactosidase activity 1.8- and 1.6-fold, respectively, relative to the DMSO control.

NaB and 5-aza C enhanced β-galactosidase staining and activity in differentiated BMEL-TAT LPCs.
BMEL A-EGFP cells undergo an epithelial to mesenchymal transition during extended culture
BMEL A-EGFP LPCs lose their epithelial phenotype after extended culture. The epithelial markers E-Cadherin and EpCAM were no longer expressed in BMEL A-EGFP after extended passaging (p57, Fig. 5, a and d), but present at p10 (Fig. 1, j and m). The opposite was true for the mesenchymal cell surface marker, vimentin. This was strongly expressed in high-passage (p57) BMEL A-EGFP (Fig. 5g), but not seen in low-passage (p10) BMEL A-EGFP (Fig. 1p).
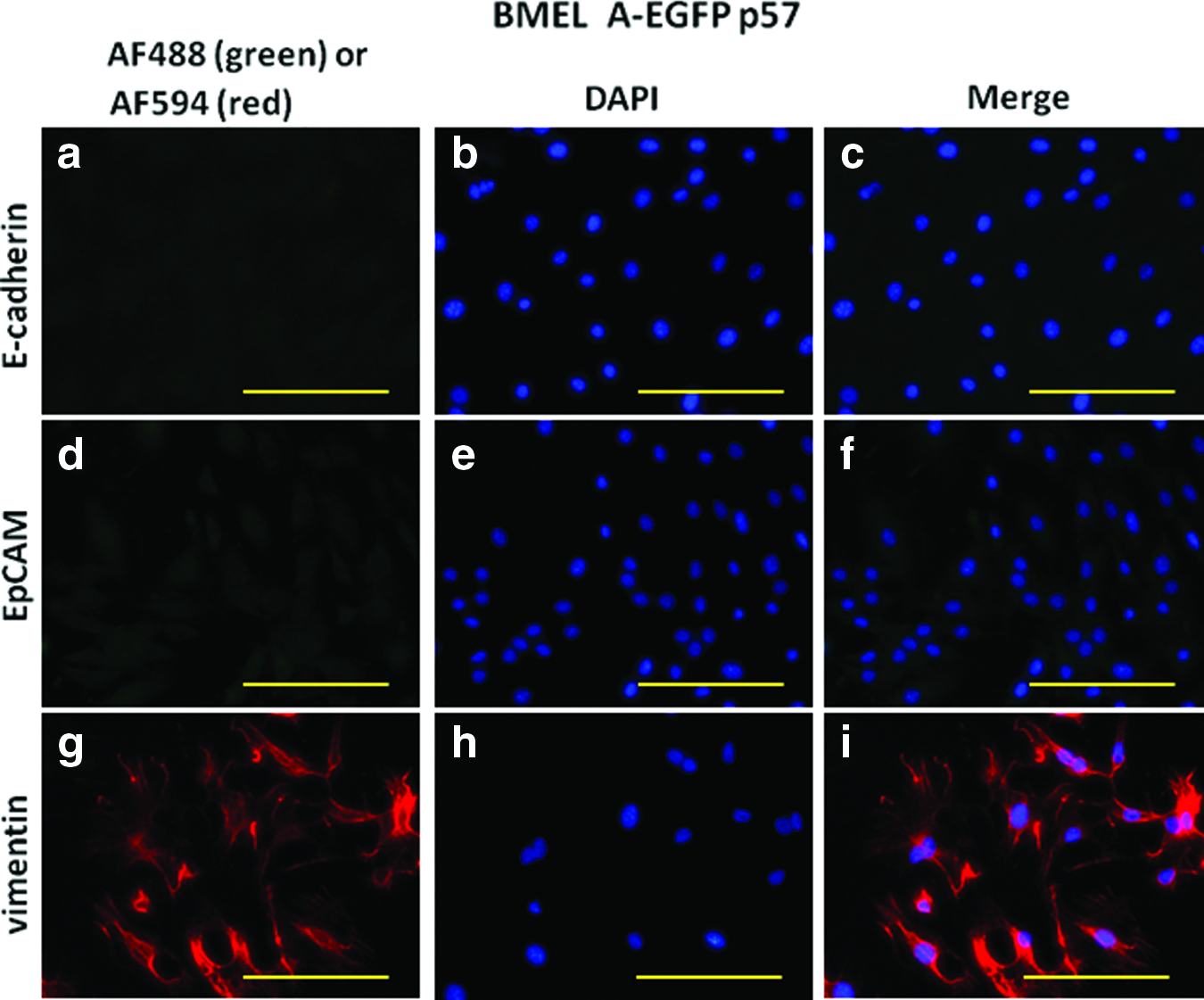
Highly passaged BMEL A-EGFP LPCs lose epithelial and gain mesenchymal cell markers. High-passage (p57) BMEL A-EGFP LPCs do not stain for epithelial markers E-Cadherin
Treatment with NaB and 5-aza C increases EGFP activity of low-passage (p10) and high-passage (p57) BMEL A-EGFP LPCs
To ascertain whether a constitutive reporter in LPCs responds in a similar manner to a reporter that is differentiation dependent, the fluorescence of BMEL A-EGFP (p10 and p57) cells treated with NaB or 5-aza C was analyzed each day for 3 days. This showed an increase in average pixel intensity for p10 BMEL A-EGFP cells from day 0 to 3 when treated with NaB (from 1.5 to 5.6) and with 5-aza C (from 1.2 to 6.5) (Fig. 6A). Extensively passaged cells (p57) showed an average pixel intensity increase from 0.1 to 1.1, 0.4 to 1, and 0 to 0.7 after treatment with NaB, 5-aza C, and DMSO, respectively (Fig. 6B).
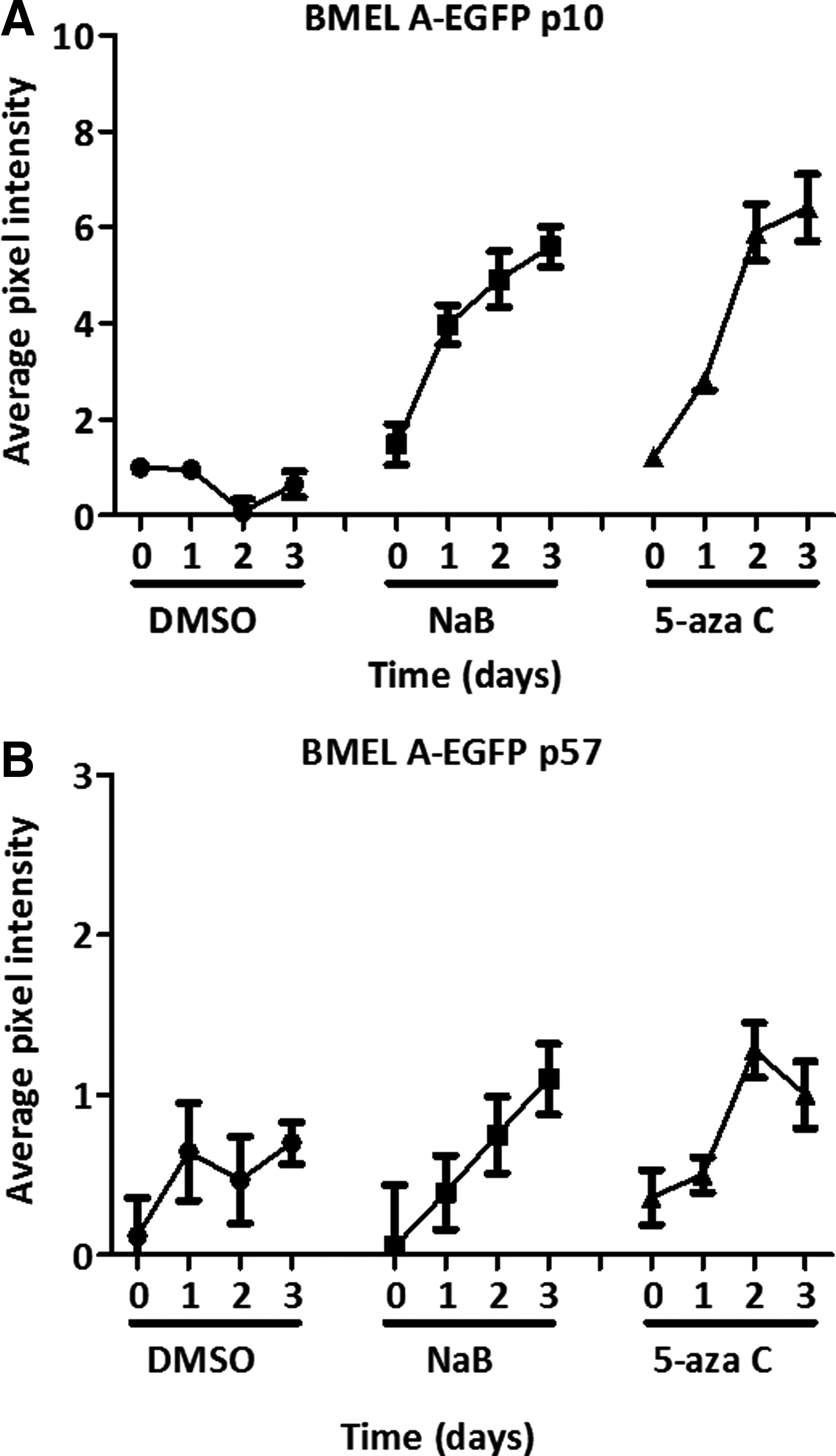
EGFP fluorescence in BMEL A-EGFP LPCs increases following treatment with NaB and 5-aza C. Shown are the relative EGFP fluorescent intensities of BMEL A-EGFP at p10
Flow cytometry-confirmed treatment with NaB or 5-aza C enhanced the activity of the EGFP reporter in the BMEL A-EGFP LPCs. Treatment with NaB or 5-aza C increased fluorescence by 4.7- and 4.4-fold, respectively, relative to LPCs treated with DMSO. There was no shift in EGFP expression in DMSO-treated LPCs relative to the medium alone (Fig. 7).
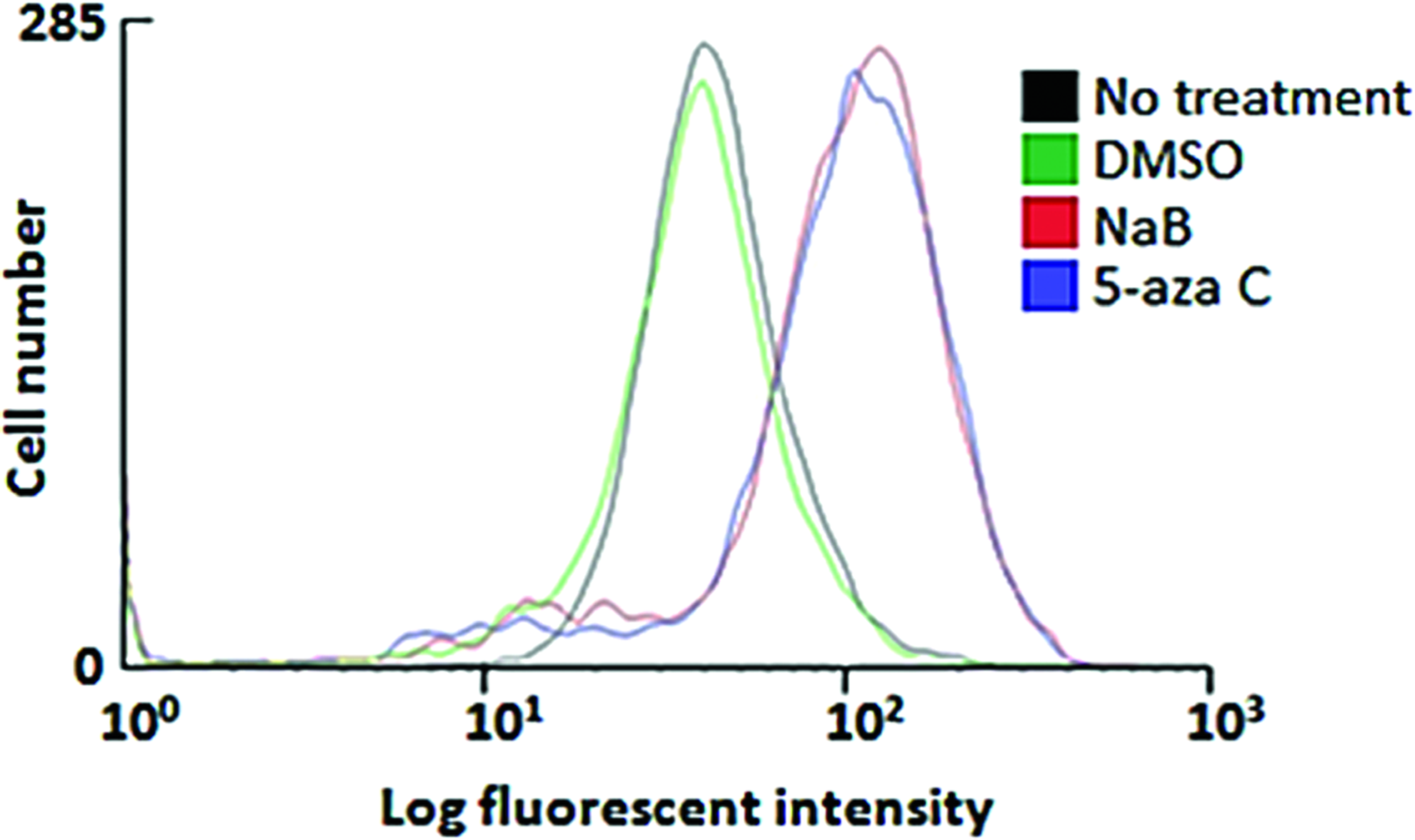
NaB or 5-aza C increases in EGFP expression in BMEL A-EGFP LPCs. EGFP levels were analyzed by flow cytometry following no treatment (black) or after 3 days of treatment with DMSO (control) (green), NaB (red), or 5-aza C (blue), respectively. For each sample, over 9000 events were acquired in the list mode and analyzed with the CellQuest Pro software (Becton Dickinson). Color images available online at
Discussion
The current study demonstrates that epigenetic mechanisms downregulate the expression of transgenes in two independent LPC lines during culture. This diminishes the utility of the cell lines for tracking or lineage evaluation experiments or, in extreme cases, renders them unusable. We have shown that two chemical epigenetic modulators, NaB and 5-aza C, enhance transgene expression in these mouse LPC lines. Our findings highlight a number of important principles associated with maintaining transgene expression in stem and progenitor cells.
In BMEL-TAT LPCs, a decrease in β-galactosidase activity accompanies increased passage number. There are two possible explanations; either epigenetic changes have suppressed the ability of the cells to differentiate or they have downregulated expression of the reporter. We suggest the latter is more likely for two reasons. First, the NaB and 5-aza C treatments increased expression of both the constitutive (EGFP) and the differentiation (β-galactosidase) reporter. Second, the level of reporter expression was increased on a per cell basis and not numbers of positive cells. These findings suggest that the epigenetic changes affect the level of transgene expression and not the ability of the cell to express the transgene.
The extensively cultured BMEL A-EGFP (p57) LPCs increased reporter fluorescent levels following treatment with NaB or 5-aza C, although the increase was not as large as that seen with BMEL-TAT LPCs or the lower passage BMEL A-EGFP (p10) LPCs. This suggests that they may be more resistant to epigenetic modulators to reverse silencing of the reporter gene. The BMEL A-EGFP LPCs undergo an epithelial to mesenchymal-like transition after extensive passaging that may render them more resistant to epigenetic modulation. This highlights the need to avoid epigenetic changes in cell lines carrying reporter genes of interest or risk losing their utility.
Conclusion
This study shows that epigenetic mechanisms diminish reporter gene expression in LPCs and, importantly, this can be reversed. This is especially significant for the BMEL-TAT LPCs as this transgenic mouse is no longer available to derive new cell lines. Treatment with NaB or 5-aza C enhances transgene expression in a subpopulation that allows for their selection and use in future experiments. These findings suggest that epigenetic mechanisms may operate during the maintenance of stem and progenitor cell lines and highlight the need to adopt culture conditions that minimize or eliminate these deleterious effects. If this is not possible, then treatment with epigenetic modifiers as we have shown here may restore sufficient reporter activity and utility of the cell line.
Footnotes
Acknowledgments
This research has received grant support from the National Health and Medical Research Council of Australia and the Cancer Council Western Australia. Additional funding was provided by the Ride to Conquer Cancer from the Harry Perkins Institute of Medical Research. We thank Heidi Peters of the University of Melbourne for supplying the BMEL A-EGFP cell line.
Disclosure Statement
No competing financial interests exist.