
Abstract
Large, deep full-thickness skin wounds from high-graded burns or trauma are not able to reepithelialize sufficiently, resulting in scar formation, mobility limitations, and cosmetic deformities. In this study, in vitro-constructed tissue replacements are needed. Furthermore, such full-skin equivalents would be helpful as in vivo-like test systems for toxicity, cosmetic, and pharmaceutical testing. Up to date, no skin equivalent is available containing the underlying subcutaneous fatty tissue. In this study, we composed a full-skin equivalent and evaluated three different media for the coculture of mature adipocytes, fibroblasts, and keratinocytes. Therefore, adipocyte medium was supplemented with ascorbyl-2-phosphate and calcium chloride, which are important for successful epidermal stratification (Air medium). This medium was further supplemented with two commercially available factor combinations often used for the in vitro culture of keratinocytes (Air-HKGS and Air-KGM medium). We showed that in all media, keratinocytes differentiated successfully to build a stratified epidermal layer and expressed cytokeratin 10 and 14. Perilipin A-positive adipocytes could be found in all tissue models for up to 14 days, whereas adipocytes in the Air-HKGS and Air-KGM medium seemed to be smaller. Adipocytes in all tissue models were able to release adipocyte-specific factors, whereas the supplementation of keratinocyte-specific factors had a slightly negative effect on adipocyte functionality. The permeability of the epidermis of all models was comparable since they were able to withstand a deep penetration of cytotoxic Triton X in the same manner. Taken together, we were able to compose functional three-layered full-skin equivalents by using the Air medium.
Introduction
P
These skin equivalents should contain fatty tissues with a dermis and epidermal layer for aesthetic and functional reconstruction. Up to date, autografts, like free flaps, are used, which have several limitations like availability of sufficient donor sites, donor site morbidity, and scar formation. 1 New alternatives are approached in tissue engineering. 2 Several strategies are available to allow sufficient differentiation of keratinocytes to build a stratified epidermal layer in a dermal–epidermal skin equivalent.3,4 Furthermore, for the treatment of second- and third-degree wounds, several dermis and epidermis substitutes are already in the market, which are used with cells (e.g., Apligraf®, Dermagraft®, EpidermFT™) or without cells (e.g., Biobrane®, Integra®), and are reviewed in the literature. 5 These substitutes are neglecting the subdermal fatty tissue, which is needed to cure fourth-degree wounds, where not only the dermis and epidermis but also the subcutaneous tissue is affected.
Physiologically, the adipose tissue is important for mechanical support, heat isolation, as an energy reservoir, and an endocrine organ. Therefore, first efforts have been made in the composition of three-layered tissue constructs.6–9 For the composition of fatty tissue, mainly stem cells were differentiated into the adipogenic lineage, which is very time-, material-, and cost-intensive. These results are not yet satisfactory since the subdermal layer lacks a sufficient amount of functional adipocytes. An appropriate culture medium is not available yet to allow simultaneous differentiation of preadipocytes to form adipocytes as well as keratinocytes to build up an epidermal layer. Alternatively, mature adipocytes could be used for the composition of a functional subcutaneous layer. They have promising properties since they can be isolated in high quantities from adipose tissue and are immediately ready for use and fully functional, without the need of complex differentiation processes. 10
One mainly cited drawback is the dedifferentiation of mature adipocytes under suboptimal in vitro conditions.11–13 In this study, mature adipocytes lose their typical morphology and functionality by organizing their lipids into several lipid droplets to reach a multivacuolar phenotype. Then, they undergo lipolysis, while giving rise to fibroblast-shaped, proliferative dedifferentiated fat cells (DFAT), which have multilineage character. 14 To prevent dedifferentiation, we have recently shown that mature adipocytes can be cultured in vitro in a commercially available adipocyte culture medium (AM-1; ZenBio), which was composed of factors favoring lipogenesis or inhibiting lipolysis.10,13 This medium was used in the present study as a basis to compose a full-skin culture medium.
Up to date, no coculture medium is available to add a subcutaneous layer consisting of mature adipocytes to the dermal and epidermal layer. An adipocyte medium needs to be optimized to allow cell viability and promote the differentiation and stratification of a functional epidermal layer. There are several commercially available media for the culture of dermis/epidermis equivalents, which contain factors like hydrocortisone or transferrin among others. These factors are further known to activate lipolysis or inhibit lipogenesis in adipocytes.15,16 Therefore, the aim of this study was to find optimal conditions for the composition and culture of functional three-layered full-skin equivalents, which inhibit adipocyte dedifferentiation and allow sufficient keratinocyte stratification.
Materials and Methods
Human tissue samples
All research was carried out in accordance with the rules for investigation of human subjects as defined in the Declaration of Helsinki. Patients gave a written agreement according to the permission of the Landesärztekammer Baden-Württemberg (F-2012-078; for normal skin from elective surgeries).
Cell isolation
Mature adipocytes were isolated from human fatty tissue of plastic surgeries received from Dr. Ziegler (Klinik Charlottenhaus, Stuttgart). The isolation was performed like previously described by us. 10 Briefly, visible blood vessels and connective tissue were removed. Adipocytes were isolated by collagenase digestion [125 U mL−1 in HBSS (Lonza) containing 3% bovine serum albumin (Sigma-Aldrich)] for 1.5 h at 37°C under agitation. The cell suspension was filtered through a 500 mm meshed sieve, centrifuged (19 g, 1 min), and washed twice with phosphate-buffered saline (PBS). Adipocytes were used immediately after isolation.
Fibroblasts were isolated from diced human foreskin. Tissues were incubated with dispase solution (2 U mL−1; Serva Electrophoresis) overnight at 37°C. Samples were then washed in PBS and epidermis was removed. Tissues were digested by collagenase (500 U mL−1; Serva Electrophoresis) for 45 min at 37°C. Cells and tissues were centrifuged at 200 g for 5 min, pellet was resuspended in 5 mL DMEM (Biochrom) and 10% FCS (Gibco), and centrifuged again. Tissue pieces were cultured until cells migrated out. For testings, fibroblasts were used in the passages 4 to 9.
Keratinocytes were isolated from the epidermis of human foreskin (Dr. Yurrtas; Stuttgart) by incubation with 0.05% trypsin/EDTA (Life Technologies) for 5 min at 37°C. The reaction was stopped by the addition of FCS (Gibco). Cells were filtered through a 100 μm mesh and centrifuged at 200 g for 10 min. Pellet was resuspended in the keratinocyte growth medium (EpiLife medium with human keratinocyte growth supplement/HKGS; Life Technologies) and seeded in cell culture flasks. All culture media used in this study were supplemented with penicillin/streptomycin as antibiotics. In this study, keratinocytes were used in passage 3.
Composition of full-skin equivalents and adipose tissue models
Full-skin equivalents were composed in a collagen type I hydrogel (9 mg mL−1 from rat tail, kindly provided by Fraunhofer IGB) in 24-well plate inserts (Brand). A neutralization buffer consisting of 10 × DMEM/Ham's F-12 (Biochrom) and 50 mM NaOH in Aqua dest (1:1) with 0.2 M NaHCO3 and 0.225 M HEPES (Roth) was prepared. For the subcutaneous layer, the collagen gel was mixed with the packed freshly isolated mature adipocytes (∼1.07 × 105 cells per hydrogel) and the neutralization buffer in a ratio of 4:4:1. Two hundred microliters of the mixture was pipetted into each well and gelled for 20 min at 37°C. For the dermal layer, fibroblasts were trypsinized and resuspended in the fibroblast growth medium. One portion neutralization buffer was mixed with four portions fibroblasts in the medium and four portions collagen type I. 103 fibroblasts were seeded in 200 μL collagen solution on top of the adipose tissue layer. Hydrogels were allowed to gel for another 20 min at 37°C. Keratinocytes were obtained from the cell culture flasks by accutase incubation (Life Technologies) for 20 min at 37°C. On each fatty tissue/dermis construct, 4.9 × 105 keratinocytes per cm2 were seeded in 20 μL keratinocyte growth medium supplemented with 1.44 mM CaCl2 (Roth). Wells were filled with 1 mL test medium. After 2 h, keratinocytes were attached and 0.5 mL test medium was added to allow airlift conditions for keratinocytes.
Full-skin constructs and epidermis models were cultured for up to 2 weeks in three different media compositions. The adipocyte medium AM-1 (ZenBio) containing 1.44 mM CaCl2 and 73 μg mL−1 ascorbyl-2-phosphate (Sigma-Aldrich) was used as a basal medium ( = Air). To fabricate two further test media, this medium was supplemented with two supplement packs normally used for the in vitro culture of keratinocytes: with HKGS and called Air-HKGS or with the keratinocyte growth medium supplement without the supplied CaCl2 (KGM; PromoCell) and named Air-KGM. As a control, epidermis models were cultured in the keratinocyte growth medium supplemented with 1.44 mM CaCl2 and 73 μg mL−1 ascorbyl-2-phosphate. Adipose tissue equivalents consisting of mature adipocytes in collagen type I were prepared similar to the adipose tissue layer in the full-skin equivalents and cultured in AM-1. Briefly, mature adipocytes, collagen type I hydrogel, and the neutralization buffer were mixed in a 4:4:1 ratio and 200 μL was pipetted in an insert of a 24-well plate. After gelation, cells were cultured submerged in the three culture media.
Hemalaun–Eosin staining and antibody staining
Hydrogels were fixed using Bouin's fixative (Sigma-Aldrich) for 1 h and watered with tap water for several hours. Gels were embedded into paraffin and sections of 5 μm were prepared.
Hemalaun–eosin staining was performed according to a standard staining procedure. Hemalaun stains the cell nuclei blue, and eosin interacts with eosinophilic structures like the cytoplasm for it to be shown in red.
For antibody staining, deparaffinization was done according to a standard protocol. Sections were heat demasked with a target retrieval solution pH 9 (Dako) for 20 min in a preheated steamer (Braun). Samples were permeabilized with Triton X-100 for 10 min and then blocked with 3% BSA in PBS+ for 30 min. Primary antibody (Perilipin A: 1:200; Sigma-Aldrich), cytokeratin 10 (1:500), and cytokeratin 14 (1:600; both Santa Cruz) were diluted with an antibody diluent (Dako) and incubated overnight at 4°C. For detection, a peroxidase-based system from Dako (EnVision+System-HRP) was applied. A secondary antibody conjugated with an HRP-labeled polymer (Dako) was used and slides were incubated for 30 min at room temperature. The staining reaction was performed with diaminobencidine (DAB, DCS) and slides were counterstained with Mayer's hematoxylin (Dako). The sections were covered with Aquatex (Merck Millipore) and a coverslip and analyzed with a light microscope (Zeiss).
Measure of adipokine release by Bio-Plex assays
To follow the cytokine response of skin equivalent samples cultured in different media compositions compared to adipocytes alone, Bio-Plex assays (Bio-Plex Pro™ Human Diabetes 10 + 2; Bio RAD) for the detection of fat tissue-related markers were carried out. Bio-Plex Assays enable the quantification of multiple protein biomarkers in a single well of a 96-well plate. Briefly, cell culture supernatants were collected on day 14 (72 h after the last medium exchange) and 50 μL samples were evaluated according to the manufacturer's instructions. Each experiment was performed in duplicate. The concentration of adipsin, leptin, visfatin, and PAI-1 was calculated using a standard curve provided by the Bio-Plex Manager Software (Bio RAD).
Determination of the epidermal barrier function
The stratum corneum is supposed to withstand a fast penetration of cytotoxic chemicals. 17 This ability was measured by treatment with 1% Triton X-100 (Sigma-Aldrich).
For each time point (0, 3 and 5 h), three tissue models were incubated airlift in 1.2 mL medium. Each keratinocyte layer was covered with a filter paper soaked in 30 μL Triton X-100 and cultured at 37°C. The filter paper was then removed and tissue models were washed thrice in PBS. To measure cell viability, 1 mg mL−1 MTT (Promega) was added and incubated for 2 h at 37°C. Extraction of the formazan salt was performed by addition of isopropanol and incubation overnight at 4°C. The extinction of 100 μL of each sample was measured at 578 nm. The values were normalized to the viability of untreated tissue models.
Statistics
All experiments were repeatedly performed, successively using cells from at least three different donors. Data were compared by one-way analysis of variance (ANOVA) with repeated measurements and a Tukey post hoc test using Origin Pro 8.5 G. Statistic significances were stated as *p ≤ 0.05.
Results
Full-skin equivalents are morphologically similar to native human skin
The potential of the tested media to supply the full-skin equivalents sufficiently with the adequate nutrients and growth factors, these models were cultured for up to 14 days. Samples were analyzed by Hemalaun–Eosin staining and compared to native human skin (Fig. 1).
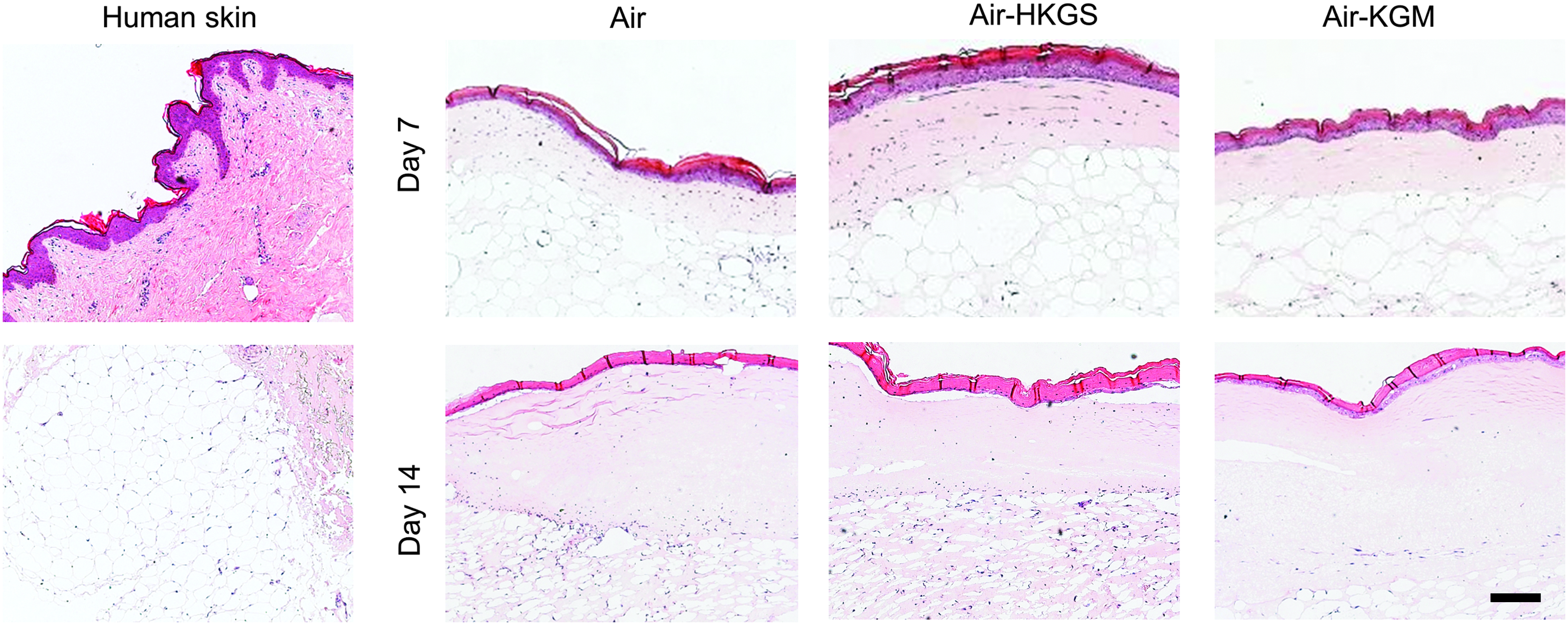
Hemalaun–Eosin staining of full-skin equivalents in several culture media after 7 and 14 days. Cell nuclei were stained with Hemalaun in blue. Eosin interacted with eosinophilic structures like the cytoplasm and showed them in red. Scale bar: 200 μm. Color images available online at
Native full skin consisted of a stratified epidermal layer, an extracellular matrix-rich dermis, and the underlying fatty tissue, mainly containing mature adipocytes giving it the typical lattice structure. Similar tissue morphology was found in the artificial full-skin equivalents, independent of the culture medium on day 7 and 14. In all samples, keratinocytes differentiated to build a cornified epidermis, whereas on day 14, a thinner and more disintegrated epidermis is seen. In all setups, fibroblasts could be found in the dermis. The subcutis showed the typical lattice structure, which could also be seen of fatty tissue in native skin.
Figure 2 shows a higher magnification of the epidermis of full-skin equivalents cultured for up to 14 days in the three test media. The epidermis of native human skin consisted of a basal layer with prismatic cells, which were overlaid by several layers of partly roundish and elongated cells. The epidermis was terminated by a thick stratified layer of keratinocytes. On day 7, the epidermis of full-skin equivalents cultured in the three culture media showed a basal layer containing roundish and prismatic-shaped cells similar to the native epidermis. This layer was covered by several layers of elongated cells and then topped with a stratified corneum. On day 14, the epidermis of tissue constructs cultured in the three culture media was left with a disintegrated epidermis containing only a very thin basal layer with partly roundish and elongated cells. This layer was covered by a thin layer of elongated cells and then terminated by a thick stratum corneum.
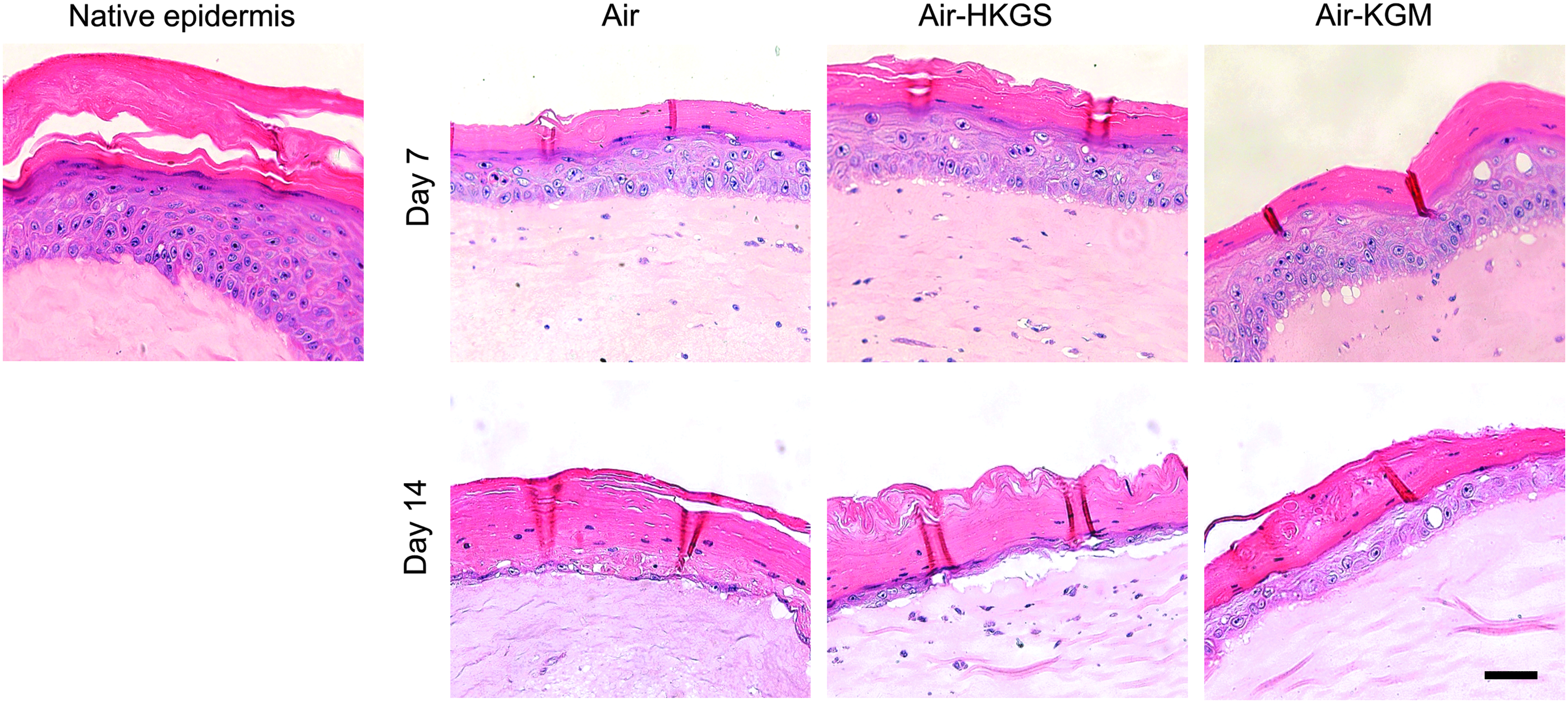
Hemalaun–Eosin staining of the epidermis of full-skin equivalents in several culture media after 7 and 14 days. Cell nuclei were stained with Hemalaun in blue. Eosin interacted with eosinophilic structures like the cytoplasm and showed them in red. Scale bar: 50 μm. Color images available online at
The expression of cell-specific markers was analyzed on day 7 and 14. Therefore, epidermis of skin equivalents was stained with both cytokeratin 10 and cytokeratin 14 (Fig. 3). Adipose tissue was analyzed using perilipin A staining (Fig. 4).
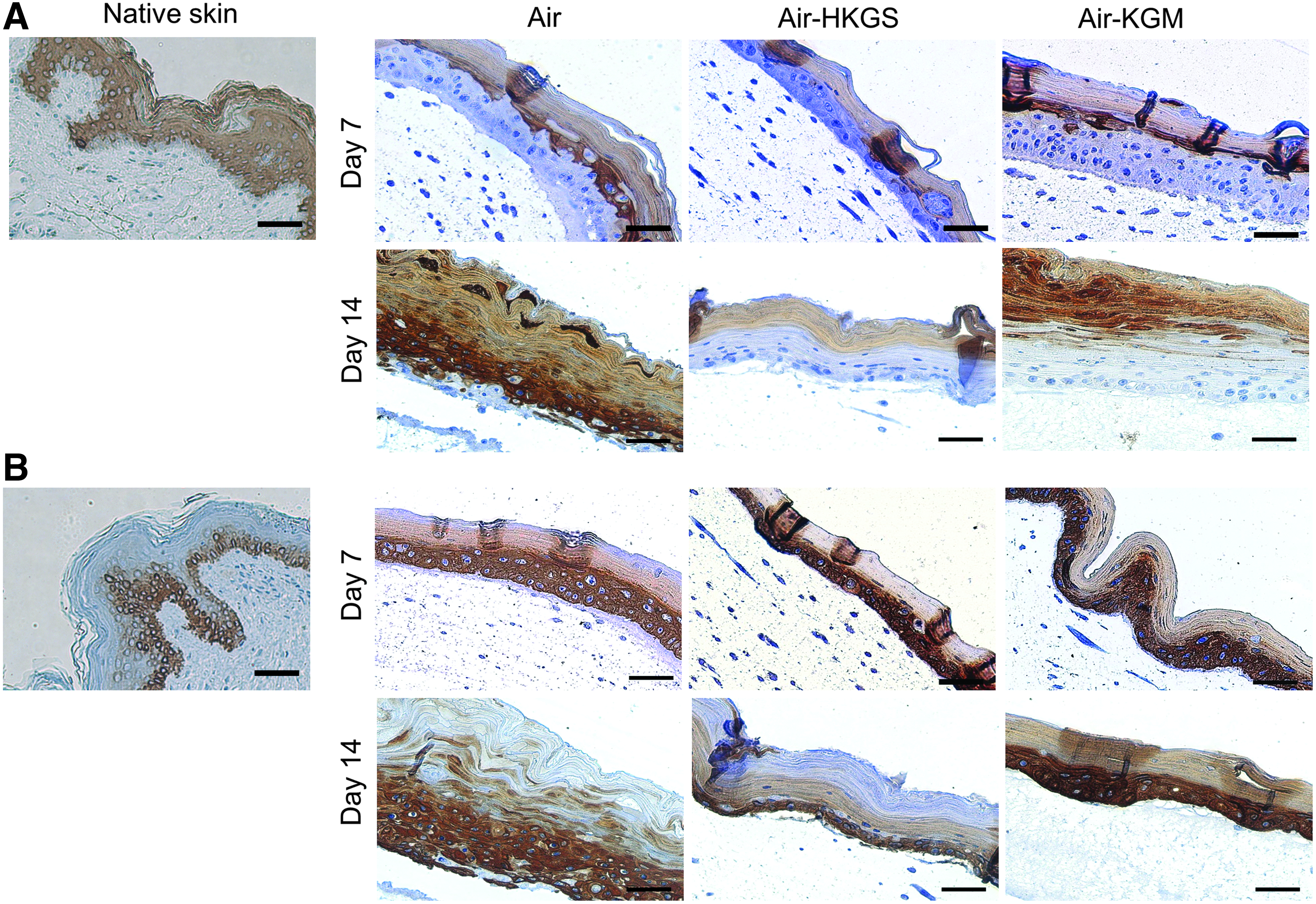
Cytokeratin 10 and 14 expression of native skin and full-skin equivalents on day 7 and 14.

Perilipin A expression of adipose tissue and full-skin equivalents on day 7 and 14. Perilipin A is shown in brown, nuclei are stained in blue. Scale bar: 50 μm. Color images available online at
All skin equivalents expressed cytokeratin 10 in the stratum corneum on day 7 and 14, which was similar to native skin tissue. Cytokeratin 14 was found in the epidermal basal layer in native tissue, whereas cytokeratin 14 could be shown in the basal layer and suprabasal layers in the epidermis of all skin tissue equivalents on both evaluated time points. Cells cultured in Air-KGM expressed cytokeratin 14 also in the stratum corneum. As shown in Figure 4, perilipin A surrounded the lipid vacuole in mature adipocytes in native tissue as well as adipocytes in all evaluated skin models on day 7 and 14. Smaller roundish cells could be found in Air-HKGS and Air-KGM in the adipose tissue on day 14, which are possibly dedifferentiated fat cells. In all samples, a few perilipin A-negative cells could be detected.
Full-skin equivalents release specific adipokines and withstand deep penetration of a cytotoxic substance
The supernatants of full-skin equivalents and mature adipocytes alone were analyzed on day 14 for the release of four adipokines. The results are shown in Figure 5.
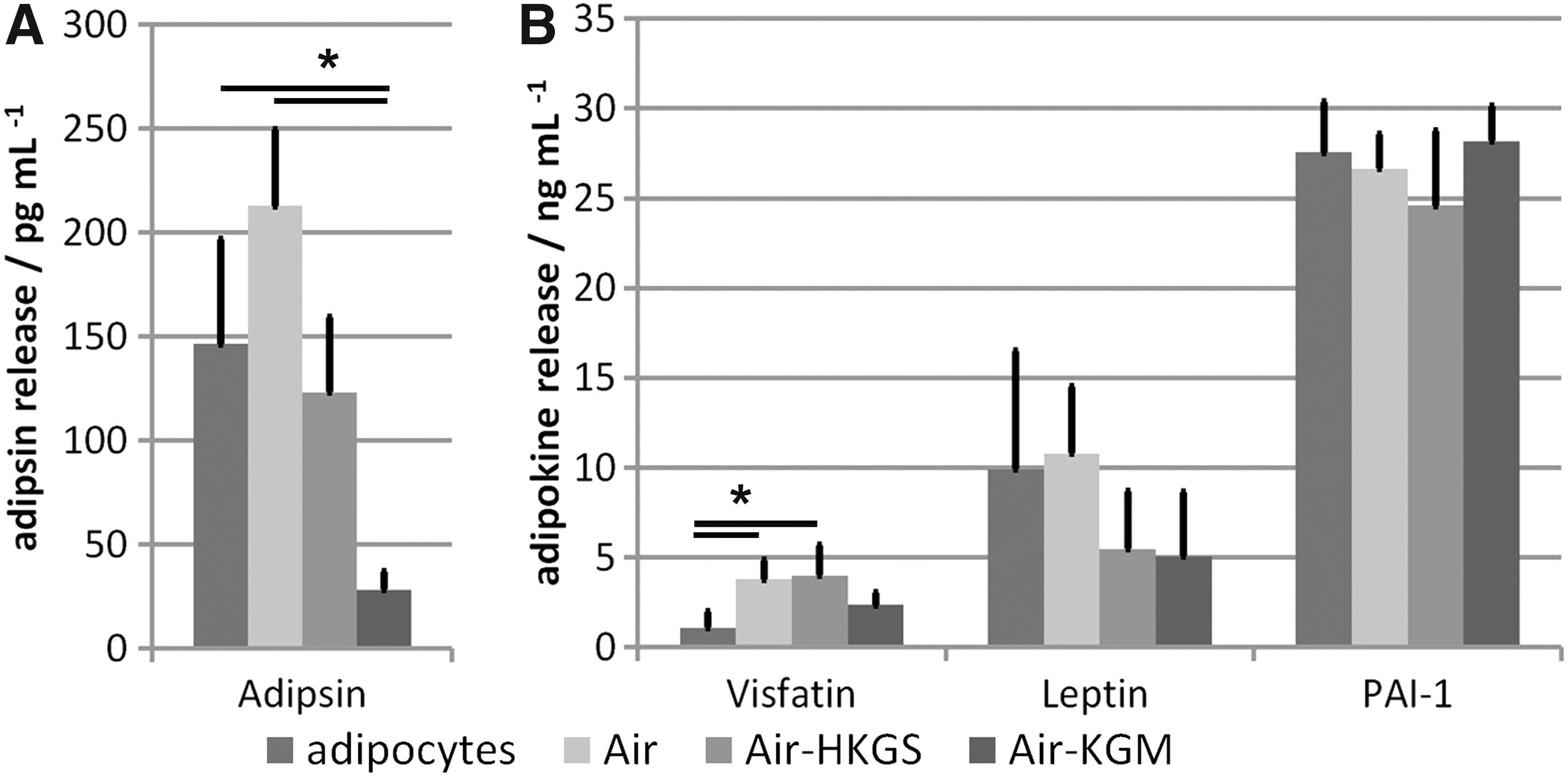
Adipokine release of full-skin equivalents holds in several media compositions on day 14.
Adipsin was released from mature adipocytes and the adipocytes cocultured in all full-skin equivalents, whereas the value was significantly lower when cultured in Air-KGM with 28 ± 8.8 pg mL−1. Mature adipocytes released 146 ± 50.5 pg mL−1 and adipocytes from full-skin equivalents in Air and Air-HKGS reached values of 213 ± 36.6 and 123 ± 36.1 pg mL−1, respectively. Visfatin was significantly less produced by adipose tissue equivalents with 1 ± 0.9 ng mL−1 compared to the full-skin tissue equivalents cultured in Air or Air-HKGS with 4 ± 1.1 and 4 ± 1.7 ng mL−1, respectively. Adipocytes in full-skin equivalents cultured in Air-KGM released 2.37 ± 0.7 ng mL−1 visfatin. A similar amount of leptin was released by mature adipocytes (10 ± 6.6 ng mL−1) compared to full-skin equivalents cultured in Air (11 ± 3.7 ng mL−1). Cells cultured in Air-HKGS and Air-KGM showed a release of 6 ± 3.2 and 5 ± 3.6 ng mL−1, respectively. A comparable amount of PAI-1 was released by adipocytes alone compared to the full-skin equivalents cultured in Air, Air-HKGS, and Air-KGM with values of 28 ± 2.8, 27 ± 1.9, 25 ± 4.2, and 28 ± 2.0 ng mL−1, respectively.
To determine the ability of the epidermis to withstand cytotoxic substances, the full-skin equivalents were treated with 1% Triton X-100. The tissue models were overlaid with a filter paper soaked in Triton X and incubated for up to 5 h. Cell viability was then measured with an MTT assay, nontreated skin models were set as 100%. Results are displayed in Figure 6. We were able to show that the epidermis of all full-skin equivalents cultured in several media showed a similar diffusion time of Triton X on day 7 and 14, and cells showed a viability higher than 70% for up to 5 h of incubation.

Epidermal barrier function.
On day 7, the viability of cells after 3 or 5 h of Triton X incubation was above 80% in all tested media (Fig. 6A). On day 14, after 3 h of incubation, cell viability dropped to 70% ± 13.1%, 76% ± 11.6%, and 82% ± 10.0% in Air, Air-HKGS, and Air-KGM, respectively. Cell viability in all culture media stayed constant at this level for up to 5 h (Fig. 6B).
Discussion
The treatment of large-scale skin defects also affecting subcutaneous fatty tissue is still a challenging goal. In this study, full-skin tissue engineering is a promising approach. It is still unknown on how to coculture the main actors; mature adipocytes, fibroblasts, and keratinocytes in vitro to compose a functional three-layered full-skin equivalent. In this study, we evaluated three different media for the coculture of these cells and their ability to supply a functional full-skin equivalent.
First studies in the coculture of preadipocytes with fibroblasts and keratinocytes have been performed in the last years.6–9,18 Aoki et al. cocultured preadipocytes or fibroblasts with keratinocytes in DMEM/Ham's F-12 and 15% FCS. They have shown that preadipocytes and fibroblasts increased the stratification of keratinocytes, whereas keratinocytes alone built a thinner epidermal layer. 7 Comparable to this study, Ataç et al. have added native fatty tissue to a dermis/epidermis model and cultured it with DMEM/Ham's F-12 and 10% FCS. 18 In addition, they added adipocyte-specific factors like insulin and hydrocortisone to the culture medium. They have seen that the adipose tissue had a stimulating effect on keratinocyte proliferation in the basal layer. Increased cell apoptosis in the adipose tissue was identified in statically cultured skin equivalents compared to dynamically cultured ones. In our study, we also detected a stratified epidermis after 14 days when keratinocytes were cocultured with mature adipocytes and fibroblasts. However, opposing to these studies, we have recently shown that a culture medium with high FCS levels favored dedifferentiation of mature adipocytes instead of keeping their physiologic phenotype. 13 Furthermore, our data showed that the addition of adipocyte-specific factors, like insulin or biotin, were beneficial for adipocyte culture.
To compose full-skin equivalents, Bellas et al. predifferentiated ASCs into the adipogenic lineage and combined this adipose tissue with a dermal/epidermal layer by using collagen glue. They have seen the best outcome when mixing an adipocyte induction medium with an epidermal medium (1:1), whereas some adipocyte markers still decreased over time. 6 Opposing that, Delort et al. have cocultured preadipocytes, fibroblasts, and keratinocytes in a similar mix-medium, but could not detect preadipocyte differentiation. 8 Monfort et al. predifferentiated their stem cells and added the dermal/epidermal layer while culturing the whole construct in an adipocyte medium. 9 They were able to find a stratified epidermis, whereas the amount of adipocytes was low.
Based on these data, our aim was to compose a medium for the coculture of mature adipocytes, fibroblasts, and keratinocytes, which prevents adipocyte dedifferentiation and allows keratinocyte differentiation at the same time.
Full-skin equivalents are morphologically similar to native human skin
When coculturing keratinocytes with adipocytes and fibroblasts, tissue morphology similar to that of native skin was achieved on day 7 and 14. Sugihara et al. have shown before that keratinocytes seeded on a fatty tissue equivalent proliferated extensively and built a thick epidermal layer. Opposing to that, fibroblast proliferation was inhibited by mature adipocytes. 19 In our study, epidermal differentiation could be detected and we did not find an undergrowth of fibroblasts. In our study, the epidermis of full-skin equivalents cultured for 7 days was similar to that of native tissue. However, on day 14, in all culture media, the epidermis started to disintegrate. We hypothesize that the factors responsible for the maintenance of the epidermis are not available in appropriate amounts. Therefore, we propose that a culture time of 7 days is already sufficient to compose a full-skin equivalent morphologically similar to native skin tissue.
Tissue-specific markers were expressed in all tissue equivalents to different levels on day 7 and 14. In native epidermis, cytokeratin 14 is expressed in the basal layer and cytokeratin 10 in the spinous layer. Expression of cytokeratin 10 could be found in the tissue models cultured in Air, Air-HKGS, and Air-KGM. However, cytokeratin 14 showed an expression also in untypical layers. This cytokeratin 14 expression could be explained by the shortened maturation of the stratum corneum in vitro (2 weeks). Perilipin A expression could be found in the adipose tissue of all full-skin equivalents. Still, adipocyte dedifferentiation in the Air-HKGS and Air-KGM medium needs to be further evaluated.
In summary, the addition of keratinocyte-specific factors, which were supplemented in Air-HKGS and Air-KGM, did not have any influence on the epidermal stratification. Furthermore, they might have a negative impact on the differentiation status of mature adipocytes. Therefore, we propose that full-skin equivalents can be successfully cultured for at least 7 days by using the air medium.
Full-skin equivalents release specific adipokines and withstand deep penetration of a cytotoxic substance
Up to date, mainly preadipocytes were used for the composition of the fatty tissue layer in full-skin equivalents. Several authors reported insufficient adipogenic differentiation or reduction of adipogenic markers over culture time.6,8,9 In our study, we used mature adipocytes. They bear good prospects for use in adipose tissue fabrication since they can be isolated in high amounts and are immediately functional. We could detect mature adipocytes in all constructs by perilipin A staining. It was shown before that mature adipocytes release factors like adipsin, leptin, visfatin, and PAI-1.20–23 Functionality of mature adipocytes in the full-skin equivalents was measured by the release of these factors compared to adipocytes alone on day 14.
Two test media, Air-HKGS and Air-KGM, contained factors like hydrocortisone or transferrin, which are known to promote lipolysis or inhibit lipogenesis of adipocytes.16,24 We could find reduced levels of adipsin, a protein involved in the regulation of de novo triglyceride synthesis and reesterification, 25 and leptin, a hormone regulating energy balance, 26 when cells were cocultured in Air-KGM. When cells were cocultured in Air-HKGS, only reduced levels of leptin could be found. These data suggest a lower adipocyte cell number or functionality in these media. This supports our data shown in Figure 4, where adipocytes in the media Air-KGM and Air-HKGS were smaller and might be starting to dedifferentiate.
Visfatin, a cytokine also known as pre-B-cell colony-enhancing factor, is able to mimic the functions of insulin. 27 It is known that visfatin is upregulated by hypoxia, inflammation, and hyperglycemia and can be downregulated by insulin and statins. 27 We could see a higher release of visfatin from adipocytes in all full-skin equivalents compared to mature adipocytes alone. We propose that due to the larger construct size of full-skin equivalents, cells inside the adipose tissue layer might show first signs of hypoxia, which will result in a higher visfatin release. In Figure 4, we have also shown that a few perilipin A-negative adipocytes are visible. Therefore, for the composition of large tissue constructs, a vascularization system is indispensable. 28
PAI-1 is elevated in a variety of diseases, including obesity, atherothrombosis, and insulin resistance. 29 Fain et al. showed that PAI-1 was elevated in visceral adipose tissue from obese humans, whereas there was no significant difference from normal weight and obese human subjects in subcutaneous adipose tissue. 22 Opposing to that, Eriksson et al. published data saying PAI-1 in obese humans is mainly released from subcutaneous adipose tissue. 30 For our study, we used subcutaneous adipose tissue from obese patients undergoing plastic surgery, wherefore we could expect PAI-1 release from mature adipocytes. In this study, we did not see any significant difference in the PAI-1 expression of mature adipocytes alone compared to the full-skin equivalents.
To conclude, the addition of keratinocyte-specific supplements in Air-HKGS and Air-KGM has a slightly negative effect on the functionality of mature adipocytes in full-skin equivalents. To improve adipocyte survival, mature adipocytes and ASCs could be cocultured in the fatty tissue layer in further studies. Piccinno et al. (2013) and Kober et al. (2015) have shown that ASCs enhance adipocyte cell survival.31,32
Barrier function of the epidermal layer and its protection of the underlying dermis and subcutis were evaluated by Triton X application and subsequent MTT assay. According to the OECD guidelines, 17 a functional epidermis should be able to withstand or decelerate the diffusion of cytotoxic Triton X. 17 Therefore, the exposure time needed to reduce the viability of keratinocytes to 50% is determined and called ET50. For epidermis models, the lower acceptable limit was set as 4 h, whereas the upper acceptable limit was set at 8 to 10 h. 17 In our setup, we did not validate the epidermis alone, but in combination with the full-skin equivalents. It was shown that the epidermis of full-skin equivalents in all culture media was able to withstand a deep penetration of Triton X over 5 h of incubation, and around 70% to 80% of the cells stayed viable on day 7 and 14. Therefore, we can conclude that a comparable dense stratum corneum was present when cells were cultured in all three media.
Conclusion
The treatment of skin tissue defects also affecting the subcutaneous layer is still a challenging goal. Tissue-engineered full-skin equivalents, containing the underlying adipose tissue, are highly valuable for clinical use or as improved complex skin models for toxicity, cosmetic, and pharmaceutical testing. In this study, we were able to show that mature adipocytes can be successfully cocultured with fibroblasts and keratinocytes to compose a functional 3D full-skin equivalent. A culture time of 7 days is already sufficient to compose a full-skin equivalent, which is morphologically similar to native skin tissue. The Air medium is before Air-HKGS and Air-KGM since the additional supplements found in these two media are nonessential for the coculture and had no further positive effect. In further studies, these equivalents need to be validated for use as test systems and in vivo implants compared to the two-layered dermis and epidermis model. For the culture of larger constructs, a vascularization system is indispensable.
Footnotes
Acknowledgments
The authors thank Dr. Ziegler (Klinik Charlottenhaus, Stuttgart) and Dr. Yurrtas for the kind provision of human fatty tissue and skin samples from plastic surgery. A warm thank you to Silvia Kolbus–Hernandez for her extraordinary helpful support in the laboratory (Fraunhofer Institute for Interfacial Engineering and Biotechnology IGB). We further thank Brigitte Höhl (Fraunhofer IGB) for her help in all the histology issues.
We kindly acknowledge the European Commission for funding the ArtiVasc 3D project under the Seventh Framework Program (grant agreement n°263416).
Disclosure Statement
No competing financial interests exist.