
Abstract
On biocompatible implant surfaces, cellular behavior and fate of stem cells are determined not only by microenvironmental signals but also by electrochemical signals. The potential of electric fields (EFs) to stimulate bone growth and bone healing has been widely demonstrated, but the molecular mechanism linking EFs to osteogenic differentiation has remained elusive. Here we show that constant EFs triggered osteogenic induction of mesenchymal stem cells (MSCs) on a defined nanotubular TiO2 substrate. EFs stimulate the formation of plasma membrane protrusions and the transport of connexin 43 to these protrusions. Connexin 43 is required for the EF-induced lasting intracellular calcium increase, which rapidly propagates to neighboring cells by gap junctions. This enables simultaneous osteogenic induction following downstream calcineurin/CAMKII/NFAT signaling. We propose that connexin 43-mediated, EF-induced osteogenic differentiation of MSCs on a defined nanotubular titanium oxide surface may give new insight on therapeutic interventions for bone regeneration and tissue engineering approaches.
Introduction
T
Since 1959, more than 100 clinical studies and more than 30 in vitro studies were published utilizing a constant or pulsed EF or electromagnetic field for bone regeneration.9–12 Overall, the meta-analysis indicated a beneficial effect of EF application in bone repair, but the trials have shown considerable variations depending on the treatment regimen and study design, thus hampering a direct comparison and a critical evaluation of results. 9 Despite beneficial effects of EFs on bone healing, the mechanism linking between electric stimuli sensing on the cell surface and synchronized osteogenic induction in bone is poorly understood.
The aim of this study was to elucidate the mechanism of constant EF-induced mesenchymal stem cell (MSC) differentiation on biocompatible TiO2 implant surface. Recent advances in nanotechnology and materials science enabled the fabrication of well-defined, self-organized, nanoscale topography coatings on titanium-based implant materials, which support growth and differentiation of osteogenic cells in vitro and in vivo.13,14 Among the biocompatible implant materials suitable for bone repair, titanium has been widely accepted as the most favorable for osteogenic differentiation in vitro and bone regeneration in vivo. Moreover, titanium substrates with well-defined nanoscale topography allowed highly reproducible cell behavior responding to the topographic cues from the oxide surface.
Previously, we have developed vertically aligned TiO2 nanotubular layers where individual tube diameters were precisely adjustable in the range of 15–100 nm, using anodic self-ordering oxidation of titanium at different voltages. 13 We demonstrated that MSC behavior, including proliferation, migration, and differentiation, strictly responds in a diameter-dependent manner to nanoscale topography of ordered TiO2 nanotubular surfaces. In the presence of biochemical supplements (Na-ascorbate, β-glycerophosphate, and dexamethasone) for osteogenic stimulation, we found a maximum of osteogenic differentiation on 15 nm-diameter nanotubes compared to other diameters or compact TiO2. 13
In the field of bone tissue engineering using biocompatible implants, cellular behavior and cell fate of stem cells has been known to be determined not only by microenvironmental signals, such as substrate topography, soluble growth factors and cytokines, and cell–cell and cell–extracellular matrix interactions, but also by electrochemical signals. 14 So far, the efficiency of EF-induced osteogenic differentiation on nanoscale defined topographical substrate has not been reported. In the present study, we applied constant EFs under physiologically relevant current flow level to MSC on geometrically well-defined 15 nm TiO2 nanotubular surfaces without any osteogenic chemical supplement.
Previous EF studies used high voltages up to 10 V cm−1, generating a current of several mA in short-term experiments.4,5 To avoid undesired effects such as hydrolysis and accompanying pH- and ionic-gradients in long-term experiments, the present study used EF up to 0.4 V cm−1, which allowed long-term osteogenic differentiation. Furthermore, to verify how MSC behave on TiO2 nanotubular surfaces, we applied z-axis-directed EF to MSC plated on 15 nm TiO2 nanotubular layer as an anodal electrode for osteogenic differentiation.
In the present study, we show that MSC sense and react to x-y planar or z-axis-directed EFs by increased intracellular Ca2+ levels. At an initial phase of EF application, we found that connexin 43 transports to the EF-triggered membrane protrusions of the plasma membrane. Connexins, which form gap junctions and mediate cell-to-cell communication in bone, have recently been reported to also form hemichannels on the plasma membrane. 15 Connexin 43 (Cx43) is the most abundant connexin in bone and contributes to osteogenic differentiation in vitro 16 and in vivo. 17 Our data show that connexin 43 and/or gap junctions are required for the EF-induced calcium rise, and that subsequent calcium signals spreading into adjacent cells trigger simultaneous osteogenic induction of MSCs.
Materials and Methods
Nanotube formation
Titanium foils (99.6% purity; Advent Ltd.) were used for producing nanostructured surfaces. For nanostructured samples, 1 M NaH2PO4 (Sigma Aldrich) with addition of 0.125 M HF (40%; Sigma Aldrich) was used as electrolyte. Before anodization, titanium foils were cleaned by ultrasonication for 5 min, first in acetone and then in ethanol, followed by rinsing with deionized water and drying in a nitrogen stream. Anodization was carried out with potentials of 1 V at room temperature (Fig. 1A, B). All electrolytes were prepared from reagent-grade chemicals and deionized water. After anodization, the obtained samples were rinsed with deionized water and dried in a nitrogen stream. For morphological characterization of sample surfaces, a field emission scanning electron microscope (S-4800; Hitachi) was used.

Electrical field-triggered membrane protrusions contain connexin 43.
EF application devices
The setup built to deliver constant currents between planar electrodes in a physiological extracellular solution is schematically shown in Figure 1D. The reference electrode and anode/cathode were made of platinum wire and sheets, to enable a long-term use with constant currents avoiding the electrodes being damaged or corroded by the culture medium. The x-y planar EF device was installed in a six-well culture plate with 1.5 cm working distance between the anode and cathode (Fig. 1D, upper panel). Similarly, a z-axis EF device was designed (Fig. 1D, lower panel) with the MSC-loaded TiO2 nanotubular layer being the bottom electrode. For both EF devices, currents were stable with EF strength of 200 mV cm−1 for experiments lasting several weeks, as shown in the current profiles measured during 40 h of EF engagement (Fig. 1C). In a preliminary study, more than 800 mV cm−1 of long-term EF strength was shown to induce cell damage. Therefore, we used a constant EF strength of 200 or 400 mV cm−1. For short-term calcium measurement, 400 mV cm−1 of EF strength was applied on cells plated with a cell density of 5000 cm2 on nanotubes. For the long-term osteogenic differentiation, a constant EF strength of 200 mV cm−1 was applied.
Cell culture
Previously established clonal rat MSCs 13 were cultured and used for EF experiments. Clonal cells were first expanded in a stem cell medium containing 60% DMEM-LG (Life Technologies), 40% MCDB-201 (Sigma), and supplemented with 1× insulin–transferrin–selenium, 1 × linoleic acid bovine serum albumin, 100 μM ascorbic acid 2-phosphate, 100 U mL−1 of penicillin, 10 ng mL−1 EGF (Sigma), 1000 U mL−1 of streptomycin (Life Technologies), 10 ng mL−1 PDFG-BB (R&D Systems), 1000 U mL−1 of rat LIF (Millipore), and 2% fetal calf serum (FCS; Hyclone Laboratories). For EF experiments, MSC were further expanded in alpha medium containing 10% FCS, 100 U mL−1 penicillin, and 1000 U mL−1 streptomycin (Life Technologies). MSC or MC3T3-E1 cells plated on TiO2 nanotubular layers were cultivated in alpha medium containing 10% FCS throughout the EF experiments. To investigate the direct effect of EF on osteogenic differentiation of MSC, in all experiments, cells were cultured in the absence of biochemical- or growth factor-mediated stimulation. To visualize cells in situ, a stably transfected green fluorescence protein (GFP)-expressing clone was used. For osteogenic differentiation, cells were plated at a density of 10,000 cm2 and further cultivated for 8 days in the presence or absence of z-axis-directed EF. Cultivated cells were analyzed by immunocytochemistry and quantitative confocal Z-stack assay. To verify osteogenic gene expressions in short-term EF application, cells were incubated under z-axis-directed EF for 18 h and under x-y or z-axis-directed EF for 3 days compared to gene expressions in the absence of EF by RT-PCR. The culture medium was replaced every 2 days in all experiments.
Immunofluorescence and immunogold staining
For osteogenic differentiation, cells plated with a cell density of 10,000 cm2 on nanotubes were cultivated under z-axis EF for 8 days. Afterward, MSC were rinsed in PBS and fixed with 4% PFA in PBS at room temperature for 10 min. After fixation, cells were permeabilized with 0.2% Triton X-100 in PBS for 2 min, washed with PBS, and incubated with antibodies of mouse monoclonal anti-osteocalcin (Takara) and rabbit polyclonal anti-osterix (Abcam) for 1 h. Secondary antibodies were labeled with anti-rabbit Cy3 and anti-mouse Cy5 (Biosource), respectively. Cell nuclei were stained blue with DAPI (Roth). Fluorescence was monitored with a microscope (Axiophot; Carl Zeiss MicroImaging) or by confocal microscopy, using a Zeiss confocal laser scanning microscope (Model LSM 780 NLO).
For colocalization of Cx43 with a plasma membrane-targeted green fluorescent protein, MSC were transfected with a plasma membrane-targeted EGFP (a gift from Sandra Lehnert, Erlangen-Nürnberg University). Under x-y planar EF, intracellular calcium was monitored with fura-2 AM as described below. When the first increase in calcium was observed, cells were immediately fixed with 4% PFA and immunostained for Cx43 (Abcam) as described earlier. Colocalization of Cx43 and the plasma membrane-targeted EGFP was determined by a Z-stack with 2 μm slice distance on the confocal microscope.
For immunogold staining, cells were fixed 1 h after EF application with 2.5% glutaraldehyde solution (Merck) overnight at 4°C, and washed with PBS, including 0.2 mM glycine. Cells were permeabilized with 0.2% Triton X-100 in PBS. The samples were blocked with bovine serum albumin and incubated with the primary antibodies, rabbit polyclonal anti-Cx43 (Abcam) or mouse monoclonal anti-integrin β1 (Invitrogen), for 1 h. After washing with PBS, the samples were incubated with biotinylated anti-mouse or anti-rabbit IgG & M antibodies, and further incubated with streptavidin-conjugated 10 nm gold particles for 1 h, followed by washing the samples with PBS. For SEM observation, samples were rinsed in water, dehydrated in a series of acetone (60%, 70%, 80%, 90%, and 100%), and critical point dried with a critical point dryer (CPD 030; Balzers).
Calcium imaging
Cells were plated at a cell density of 5000 cm2 on TiO2 nanotubes in alpha medium (Invitrogen) containing 10% FCS and the next day cells were loaded with fura-2 AM (3 μM; Biotrend) in the presence of pluronic F-127 (0.02%) for 30 min. Cells were transferred into a HEPES-buffered extracellular solution (140 mM NaCl, 5 mM KCl, 1.25 mM CaCl2, 2 mM MgCl2, 10 mM glucose, and 10 mM HEPES, pH 7.4) and after 10 min mounted on an inverted microscope equipped with a 10× objective. Calcium was monitored by illumination by a monochromator (Polychrome V; Till Photonics) alternating between 340 ± 10 nm and 380 ± 10 nm, and fluorescence emission above 440 nm was acquired at a rate of 1 Hz by a cooled CCD camera. A 340 nm/380 nm ratio image time series was calculated after background subtraction. Calcium changes were identified in a false color image of the increase at 340 nm plus the decrease at 380 nm. Regions of interest were adapted to cells and used to derive the time course of fluorescence ratios. All experimental protocols were repeated on separate days and performed at least in triplicate. During the measurements, x-y axis-directed EF was applied at constant strength (400 mV cm−1).
To verify the source of calcium entry, we pretreated with thapsigargin 2 μM for 30 min after Fura2 staining to deplete the store-operated calcium stores. For experiments without extracellular calcium, cells were transferred into a calcium-free HEPES-buffered extracellular solution 30 min before measurement. To test the effect of gap junction inhibitors, cells were first loaded with Fura2 and then exposed to methanandamide (50 μM) or nitrophenylpropylaminobenzoic acid (NPPB, 50 μM) for 30 min before measurement of the calcium changes under EF.
To determine the time of calcium activation, an increase over 33% of the maximum calcium increase was used as an activation time point. To distinguish a continuous spread from an independent spontaneous activation, we chose 100 triples of cells. First a reference cell with no activated surrounding cells was chosen. Then, blinded to their further fate, we chose a cell adjacent to the reference cell and randomly a distant cell with no activated neighbor. The latency between activation of the reference cell and the other two cells was measured. The percentage of spontaneous activating cells without EF was compared to the fraction of activated cells 50 min after the EF onset.
Introduction of Cx43 cDNA or Cx43 siRNA into MSC on TiO2 nanotubular surfaces was performed with the K2 transfection system (Biontex), as recommended by the manufacturer. As a negative control for siRNA, a scrambled sequence of the connexin 43 siRNA target sequence was used. One day after transfection, transfected MSC were used for calcium imaging. siRNA sequences (Sigma) are as follows: Cx43 siRNA_for: CAAUUCCUCGUGCCGCAAUU and Cx43 siRNA_rev: UUGCGGCACGAGGAAUUGUU; scrambled siRNA_for: AAUUCUCCGAACGUGUCACUU and scrambled siRNA_rev: GUGACACGUUCGGAGAAUUUU.
Permeable dye spreading
To test whether gap junctional activity is affected by EF, we tested dye spreading using the permeable dye calcein and the impermeable dye DiI under EF. Unstained MSC were plated on nanotubular layer 1 day earlier at confluence. Cells labeled with DiI and calcein were dropwise plated on monolayer of unstained MSC. At 2 h after plating of stained cells, spreading of dye toward neighboring cells was observed by fluorescence microscopy.
Quantitative real-time PCR
For the detection of osteocalcin and osterix gene expressions, MSC plated at a cell density of 5000 cm2 on TiO2 nanotubular surfaces were cultivated for 3 days under EF. MSC cultivated in the absence of EF during the same period served as controls. Total RNA was extracted from MSC with the TRIzol reagent (Life technologies). Reverse transcription of RNA (1 μg) was performed with oligo(dT) or random hexamer primers and M-MuLV reverse transcriptase (Promega). The PCRs were done in triplicate for each cDNA probe using the qPCR master mix (Eurogentec) and the real-time PCR system (Applied Biosystem; TaqMan 7500) according to the manufacturer's instructions. For real-time PCR, cycling consisted of 50°C for 2 min, denaturing at 95°C for 10 min, followed by 40 cycles of 94°C for 15 s, 60°C for 60 s, and 72°C for 30 s. Primer sequences were as follows:
osteocalcin_fwd GACGAGCTAGCGGACCACAT, osteocalcin_rev CTGGAGATAGCCAAAGCTGAAG, osteocalcin_taq CTTCCAGGACGCCTACAAGCGCA; osterix_fwd TGACTGCCTGCCTAGTGTCTACA, osterix_rev TGGTGCCCGCCTTGT, osterix_taq ATGTCCCATCCCTACGGCTCCTGG; HPRT_fwd GCAGTACAGCCCCAAAATGG, HPRT_rev TCATTATAGTCAAGGGCATATCCAAC, HPRT_taq TGCAAGCTTGCTGGTGAAAAGGACCTCTC.
For RT-PCR, primer sequences were as follows:
osteocalcin_fwd CCTGGCTGCGCTCTGTCTCT, osteocalcin_rev GACATGAAGGCTTTGTCAGCTC; osterix_fwd AAGAGGTCACTCGCTCTGACG, osterix_rev ACAGAGCAGGCAAGTGAACTTCT; ALP_fwd GGTATGGGCGTCTCCACAGT, ALP_rev GCCCGTGTTGTGGTGTAGCT; Runx2_fwd CTTCATTCGCCTCACAAACAAC, Runx2_rev CCTTCTTGCAGTCTTCCTGGA; Cx43_fwd CGAGGTATCAGCACTTTTCTTTCATTGGGG, Cx43_rev GGAGATCCGCAGTCTTTGGA; GAPDH_fwd ATCACTGCCACCCAGAAGAC, GAPDH_rev ATGAGGTCCACCACCCTGTT.
Western blot analysis
Soluble proteins were extracted from MSC with lysis buffer (150 mM NaCl, 50 mM Tris, pH 7.4, 1% NP-40, 0.25% deoxycholate, 1 mM EDTA, PMSF 1 mM, NaF 1 mM, Na3VO4 1 mM, and 1 μM of aprotinin, leupeptin, pepstatin) for 10 min at 4°C. After centrifugation at 13,000 g for 20 min at 4°C, protein lysates were denatured at 90°C for 10 min, run on 10% SDS-PAGE gels, and electroblotted onto a PVDF membrane (Roti-PVDF; Roth) using standard protocols. After blocking in 5% nonfat dry milk/PBST for 2 h, the membranes were incubated overnight at 4°C with rabbit polyclonal anti-calcineurin (Cell Signaling; dilution 1:1000), anti-phospho-CAMKII (Cell Signaling; dilution 1:1000), anti-phospho-NFATc1 (Santa Cruz; dilution 1:200), and anti-Cx43 (Abcam; dilution 1:1000) antibodies, washed, incubated with the horseradish peroxidase-conjugated secondary antibody (Cell signaling; dilution 1:500), and developed for proteins using the standard ECL procedure. Equal protein loading was controlled by probing blots with an anti-GAPDH antibody (Abcam; dilution 1:20,000).
For detecting the membrane fraction of Cx43, subcellular fractionation of cell lysate was performed as described. 19 In brief, after 1 h of EF stimulation, cells were immediately lysed and centrifuged at 720 G for 5 min. To get the cytosolic and membrane fraction, the supernatant was ultracentrifuged at 100,000 G for 1 h and the supernatant provided the cytosolic fraction. The pellet was washed, resuspended, and provided the membrane fraction. After equalizing protein loading of the cytosolic fraction with an anti-GAPDH antibody, Cx43 in the membrane fraction was visualized.
ATP measurement
Cells plated at a cell density of 5000 cm2 on TiO2 nanotubes 1 day earlier were washed with PBS three times. Cells were incubated with 3 mL of PBS at 37°C for 2 h under EF. Then, 100 μL was used to measure the ATP concentration using the Luciferase Assay System (Promega) with 2.5 mg mL−1 luciferase, 1 mM luciferin, 2.5 mg mL−1 luciferase, 25 mM tricine buffer, pH 7.8, 5 mM MgSO4, 0.1 mM EDTA, and 2 mM dithiothreitol. Samples of supernatant from MSC without EF were served as controls. Relative light units were measured using the Veritas luminometer (Promega, Turner Biosystems).
Alizarin red staining
After 7 days of culture, samples were fixed with 4% paraformaldehyde, washed with distilled water, and stained with 1% alizarin red S for 1 h at room temperature to detect mineralization. For quantification, the stained cells were collected using a cell scraper and transferred into 10% acetic acid. Samples were heated at 85°C for 10 min and incubated on ice for 5 min. After centrifugation at 20,000 G for 15 min, the supernatant was transferred to a new tube. After adding 10% ammonium hydroxide to neutralize the acid, absorbance was read at 405 nm with a plate reader.
Statistical analysis
Two groups were compared with two-sided dependent or independent t-tests, multiple groups were compared by analysis of variance and the HSD post hoc test. Frequency of occurrence was tested by the chi-square test. Data are presented as mean ± SEM, and p < 0.05 was considered significant.
Results
All cellular EF experiments were performed on vertically aligned TiO2 nanotubular layers (Fig. 1B) fabricated by self-organizing anodization of TiO2 sheets (Fig. 1A).13,14 To study cell behavior under different EF directions, we used an experimental setup that controlled EF application in the x-y plane or in the z-axis (Fig. 1D), allowing prolonged exposure of MSC to static EFs for several days (Fig. 1C).
Active membrane protrusions and Cx43 transport to plasma membrane under EF
To identify whether EFs trigger the changes in cell surface structures, we examined the plasma membrane surfaces of MSC by scanning electron microscopy. Of note, we found numerous membrane protrusions (about 500 nm in length and 80 nm in width, Fig. 1F) rising from the plasma membrane (Fig. 1E and Supplementary Fig. S1A; Supplementary Data are available online at
EF triggers delayed, long-lasting [Ca2+]i increase
Calcium signaling has been known to be involved in electrotaxis.7,18 To verify whether EF stimulation can change in cytosolic-free calcium concentration ([Ca2+]i) during MSC differentiation, we analyzed [Ca2+]i changes by time-lapse calcium imaging with fura-2. We observed an EF-triggered [Ca2+]i increase (Fig. 2A), characterized by substantial latency (average 40 ± 2 min, Fig. 2B) and a long-lasting activation plateau (Supplementary Fig. S2A, C). This pattern was clearly distinct from the infrequent and short spontaneous calcium activation with an average duration of 25 s (Supplementary Fig. S2B), which was also observed in the absence of EF.

Electrical field triggers a delayed and long-lasting calcium increase.
The main calcium source for EF-induced [Ca2+]i increase was identified as extracellular calcium influx. In the absence of extracellular calcium in the medium, EF-induced [Ca2+]i rise was completely abolished, while the calcium increase under EF was slightly decreased after depletion of the endoplasmic reticulum calcium stores by thapsigargin 2 μM (−35%, p = 0.005, t-test independent samples, Fig. 2C). Next, since Cx43 is the most abundant connexin that forms gap junctions and mediates cell-to-cell communication in bone,15,16 we tested whether gap junctions contributed to the EF-induced [Ca2+]i increase. In the presence of gap junction inhibitors, methanandamide (mAEA, 50 μM) or nitrophenylpropylaminobenzoic acid (NPPB, 50 μM), the EF-induced [Ca2+]i increase was considerably reduced (−70% and −78%, both p < 0.001, Fig. 2D). Furthermore, to analyze whether Cx43 is required for EF-induced intracellular calcium rise, we performed connexin 43 overexpression or knockdown by RNA interference (Fig. 2E, F). In Cx43-overexpressing cells, the EF-triggered [Ca2+]i increase occurred with a shorter latency compared to scrambled siRNA-transfected MSC (Fig. 2E). As summarized in Figure 2G, these data strongly suggest that connexin 43 is a key factor in the EF-induced [Ca2+]i increase of MSC.
Since calcium entry through L-type calcium channels has been reported to contribute to osteogenic differentiation of MSC, 22 first we tested L-type calcium channels as a main EF-triggered calcium entry. Indeed, the EF-induced long-standing calcium increase was largely dependent on L-type calcium channel activation. The combined treatment with the L-type calcium channel antagonists D600 (10 μM) and nimodipine (3 μM) reduced the EF-induced [Ca2+]i increase (−75%, p < 0.001, Fig. 2H), indicating that L-type calcium channels carry the calcium influx under EF. Considering that L-type calcium channels are unlikely to be the direct EF sensor, we were interested in how Cx43 is involved in calcium channel activation under EF. Since Cx43 hemichannels in the plasma membrane have been suggested to release intracellular ATP into the extracellular space,22,23 inducing a calcium entry, 23 we investigated whether ATP may be responsible for inducing an EF-triggered calcium entry. Notably, extracellular application of ATP (300 μM, without EF) caused a similar pattern of long-lasting [Ca2+]i increase but faster onset of calcium activation (Fig. 2K) compared to EF stimulation. This long-lasting [Ca2+]i increase under ATP supplement was almost abolished not only by nonselective purinergic receptor antagonist suramin (500 μM, −94%) but also by L-type calcium channel antagonists, D600 (10 μM) and nimodipine (3 μM, −81%, both p < 0.001, Fig. 2I). Also, MSC exposed to EF released significantly more ATP to the extracellular space than without EF (Fig. 2L). These findings suggest that L-type calcium channel may be activated in response to Cx43-mediated ATP redistribution (Fig. 2J).
EF-triggered Cx43 transport to plasma membrane mediates intracellular calcium increase
In nonpermeabilized immunofluorescence stainings, a high amount of Cx43 was identified on the cell surface membrane following EF application (Fig. 3A), while in the absence of EF, Cx43 was rarely found on plasma membrane but found in gap junctions (Fig. 3B). In an hour after EF stimulation, a large amount of Cx43 was detected in a membrane fraction of cell lysate shown in western blot, using subcellular fractionation (Fig. 3C). To verify the temporal link between Cx43 transport to the plasma membrane and intracellular calcium increase, we investigated the cellular Cx43 localization by z-stack confocal microscopy after time-lapse calcium imaging with fura-2. When [Ca2+]i started to rise in MSC after 30 min of EF, Cx43 appeared on the plasma membrane (Fig. 3C). The localization of Cx43 on the plasma membrane is indicated by colocalization with the plasma membrane-targeted green fluorescent protein in the same Z-stack slice (Fig. 3C, arrowheads).
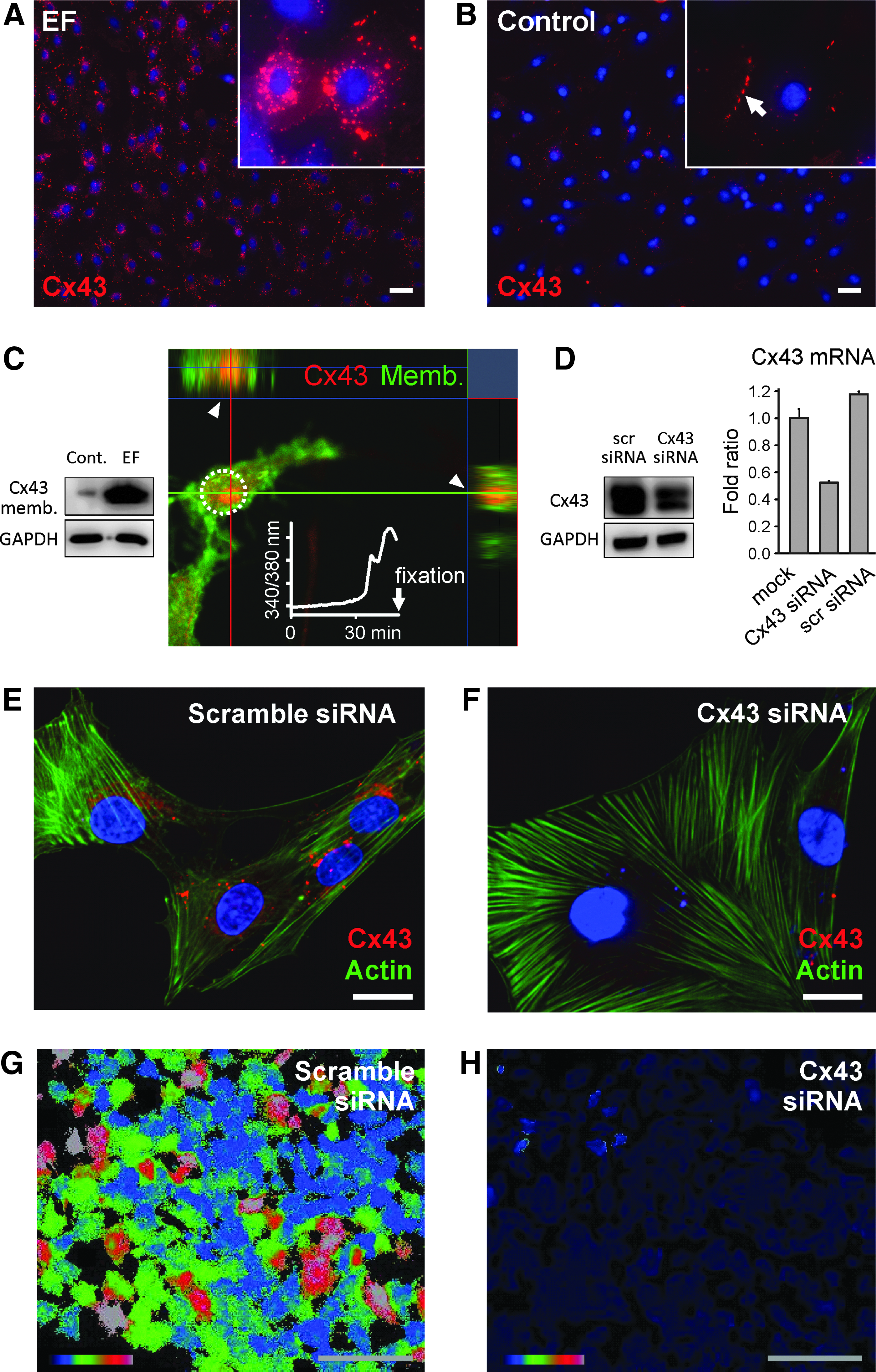
Electrical field-triggered Cx43 transport to plasma membrane.
These data indicate that the [Ca2+]i increase coincides with the Cx43 transport to plasma membrane. Furthermore, to verify whether transported Cx43 is required for EF-induced calcium activation, we performed knockdown of Cx43 by RNA interference. Efficient Cx43 knockdown was confirmed by western blot, quantitative real-time PCR (Fig. 3D), and immunostaining (Fig. 3E, F). Cx43 knockdown abolished the EF-induced intracellular calcium rise (Fig. 3H and Supplementary Video S4) in comparison to scrambled siRNA-transfected MSC as a control (Fig. 3G and Supplementary Video S3). Our data suggest that EF-triggered Cx43 transport to the plasma membrane is linked to calcium activation. Our results are in good agreement with the previous reports that Cx43 hemichannels localized to nongap junctional plasma membranes contribute to calcium oscillations. 20
Under EF, not only Cx43 transport but also membrane protrusions occurred within an hour together with calcium activation (Supplementary Fig. S3). In more than 80% of the MSC, EFs induced membrane protrusions and prolonged calcium increase (454/519 cells with EF compared to 19/1720 cells without EF, χ2 = 1785, p < 0.001). Not only MSC but also the osteoblast precursor cell line MC3T3-E1 reacted to EF similar long-lasting [Ca2+]i increase in about 20% of the cells (Supplementary Fig. S4), implying that osteogenic cells may share a common mechanism of EF-inducing calcium activation.
EF stimulates gap junctional spreading of [Ca2+]i activation
We examined whether an EF can stimulate the gap junctional intercellular spreading of [Ca2+]i. (Materials and Methods section). In the presence of the x-y planar EF, a calcein spreading from a DiI-labeled MSC toward neighboring unlabeled MSC was observed, while in the absence of EF, there is no calcein spreading from DiI-labeled MSC (Fig. 4A). Furthermore, we tested whether the EF-enhanced gap junctional activity may contribute to synchronization of EF-triggered calcium increase between adjacent cells. Intracellular calcium elevation spread to neighboring cells shortly after the first cell responded to EF (arrow in Fig. 4B panel b), and adjacent cells followed (Fig. 4B–D and Supplementary Video S5). The time of calcium elevation showed clusters of concomitant calcium elevation in neighboring cells (color-coded in Fig. 4D). Furthermore, to distinguish gap junctional calcium spreading from independent spontaneous activations, we compared the latency between activation of a reference cell and two further cells located either adjacent or distant from the reference cell. The adjacent cells were activated earlier than the distant cells, indicating a local spreading of [Ca2+]i activation to adjacent cells by EF-triggered gap junctional activation (p < 0.001, N = 100 cell triples, t-test, Fig. 4E). Such spreading activation, when affecting sufficient number of cells, frequently culminated in a wave of calcium activation with a slowly travelling wave front (Fig. 4F).
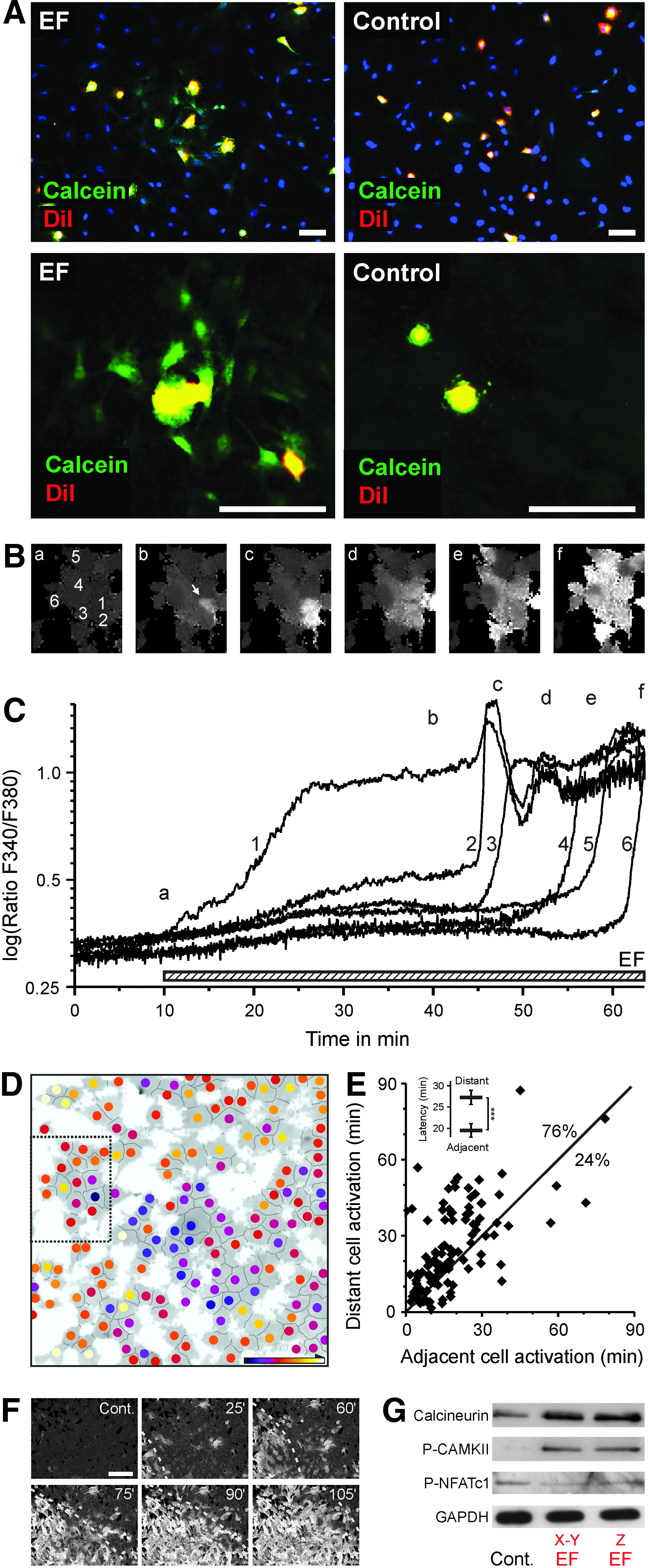
Electric field stimulates gap junctional spreading of activation to adjacent cells.
Our data showed that the EF-triggered [Ca2+]i rise further activated downstream signaling, including calcineurin/CAMKII phosphorylation and NFAT dephosphorylation, possibly leading to osteogenic gene expressions (Fig. 4G) as a potential calcium signaling pathway resulting in osteogenic induction previously reported.24–27 We hypothesize that EFs lead to Cx43 translocation and thereby ATP redistribution, which stimulates ATP receptors, causing an L-type calcium channel-based prolonged calcium influx triggering cell differentiation.
EF stimulates the osteogenic differentiation of MSC
To verify whether EF stimulates the osteogenic differentiation of MSC on a defined nanotubular layer, we applied a z-axis EF (0.2 V cm−1) for 8 days to GFP-labeled MSC loaded on TiO2 nanotubular surface serving as anode. For long-term osteogenic differentiation, MSC tolerated EF exposure at a physiologically relevant current flow level (≤ 0.4 V cm−1) without deteriorating effects. Under EF application in culture without osteogenic chemical stimulants, MSC plated as monolayer proliferated and rearranged into multilayers (Fig. 5A, left panel). In contrast, in the absence of EF, cells remained as monolayer as expected under culture condition without osteogenic stimulants (Fig. 5A, right panel). Osteogenic differentiation was indicated by strongly enhanced antibody staining for the osteoblast markers osteocalcin and osterix compared to control cultures without EF (Fig. 5B–D). Our data show that EF-triggered MSC fate decision toward osteogenic induction seemed to start at an early stage of EF application. At RNA level, the expression of alkaline phosphatase and osteocalcin was already seen after 18 h of EF application (Fig. 5E), and further upregulation of osteocalcin and osterix gene expression was detected 3 days after x-y planar or z-axis EF application (Fig. 5F). These data strongly indicate that MSC are driven toward osteogenic differentiation by a constant electrical field.
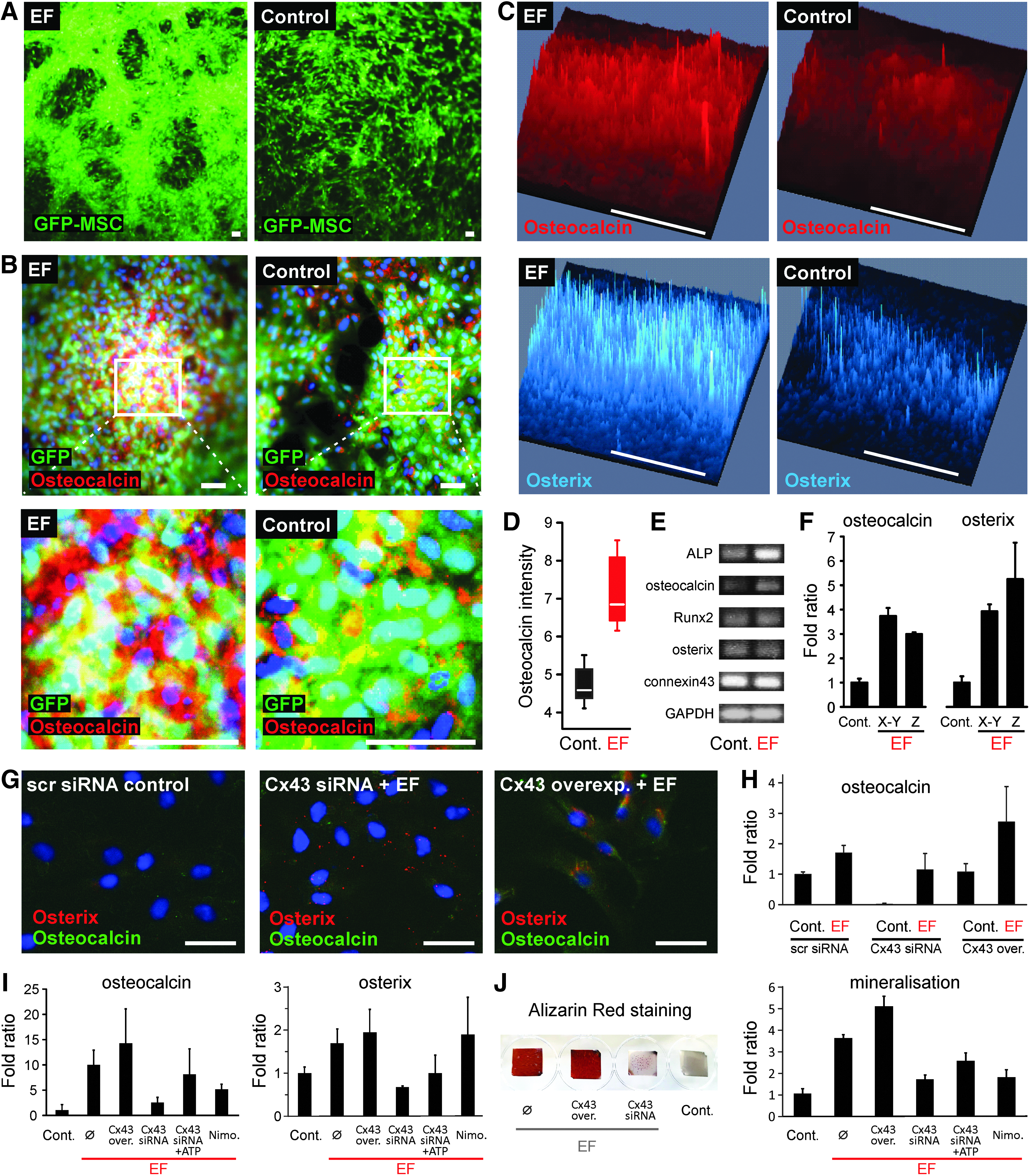
Electric field stimulates the osteogenic differentiation of MSC.
Our data showed no difference between x-y planar or z-axis EF application regarding the potential of osteogenic induction. Furthermore, to identify whether Cx43 plays a role for EF-induced MSC differentiation, we analyzed the induction of the osteogenic markers osterix and osteocalcin after 3 and 7 days of z-axis EF application in Cx43-overexpressing and Cx43-knockdown MSC (Fig. 5G–I). In immunostainings for osterix and osteocalcin (Fig. 5G), Cx43-overexpressing MSC under EF showed enhanced osterix and osteocalcin stainings. In the presence of EF, Cx43 knockdown inhibits the upregulation of osterix and osteocalcin compared to scrambled siRNA without EF. Our quantitative real-time PCR results also showed that Cx43 knockdown in the absence of EF dramatically downregulated osteocalcin expression. Under EF, induced osteocalcin expression was also decreased in Cx43 knockdown compared to scrambled siRNA-transfected MSC. While Cx43 overexpression alone did not enhance osteocalcin expression, it facilitated the EF-induced osteocalcin expression. After 7 days of EF, Cx43 knockdown dramatically downregulated osteocalcin expression. The osteocalcin expression due to Cx43-knockdown MSC was partly restored by adding 300 μM ATP (Fig. 5I). Alizarin red staining showed an EF-induced mineralization of MSC, which could be inhibited by Cx43 knockdown (Fig. 5J). These data suggest that Cx43 participates in EF-driven osteogenic induction of MSC on a TiO2 nanotubular layer (Fig. 6).
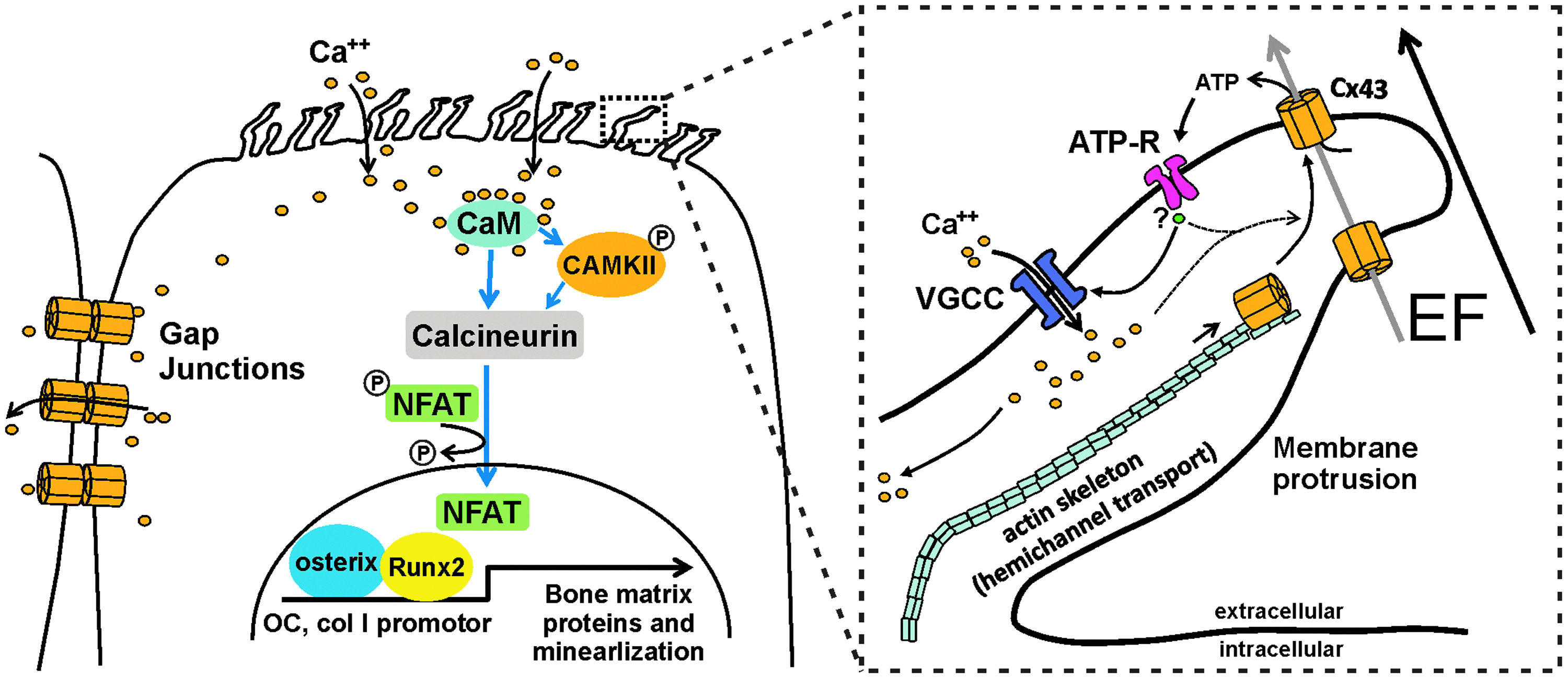
A hypothetical model of EF-triggered osteogenic differentiation. In vitro EF application facilitates MSC raising membrane protrusions with Cx43 on cell surfaces. As a potential EF sensor, EF-triggered hemichannel externalization and the activation of Cx43 permit the efflux of ATP. The latter ATP activates ATP receptors and depolarizes the cell membrane. Consequently, the activation of voltage-gated L-type calcium channels increases intracellular calcium, which may positively feedback Cx43 externalization (dashed line). Prolonged calcium elevation spreads to adjacent cells and may launch simultaneous osteogenic commitment of MSC through signaling pathways, including calmodulin (CaM)/calcineurin, and by starting the required transcriptional activation such as NFAT/osterix. Color images available online at
Discussion
Despite numerous reports on the beneficial effects of EFs on bone healing, our knowledge of the molecular and cellular mechanisms linking electric stimuli sensing on the cell surface and synchronized osteogenic induction in bone is still fragmentary. In this study, we show for the first time that application of a constant EF to MSC provokes Cx43-mediated extracellular calcium influx and gap junctional propagation of intracellular calcium increase, leading to simultaneous osteogenic differentiation.
Our study focused on how MSC react to electric stimuli. So far, under EF, calcium influx through calcium channels has been reported in electrotaxis studies.7,18 However, calcium channels are unlikely to be the primary EF sensor. Looking for an EF-sensing mediator that induces calcium channel activation, we found that MSC produce abundant membrane protrusions reacting to EF stimuli and Cx43 is actively transported to plasma membrane. Recent studies demonstrated that Cx43 units forming gap junctions or Cx43 hemichannels are transported actively on actin filaments from the endoplasmic reticulum to the plasma membrane within an hour after chemical stimulation.28,29
Our finding that EFs induce membrane protrusions with Cx43 transport on plasma membrane after a lag phase of about 30 min is consistent with the earlier report. Our observations that Cx43 with membrane protrusions appears on the plasma membrane at the onset of the intracellular calcium increase and EF-induced Ca2+ influx is strongly impaired by the Cx43 knockdown support that this transported Cx43 under EF may be a key EF-sensing mediator responsible for EF-triggered calcium influx. Although in the present study the functional role of EF-induced membrane protrusions is uncertain, the identification of Cx43 on the surface of these protrusions indicates that membrane protrusions may be rudimentary forms of canaliculi, which are abundant in bone, connecting osteoblasts and osteocytes in a dense network. How the EF instructs the organization of actin skeletons or microtubules to transport Cx43 onto the plasma membrane remains to be investigated.
Previous reports show that Cx43 hemichannels can form nongap junctional plasma membrane structures and contribute to calcium oscillations.20,25,30 Cx43 hemichannels open in response to mechanical stimulation 32 and metabolic inhibition.33–35 Our study shows the possibility that Cx43 hemichannels also open in response to electric stimuli. Open unopposed hemichannels allow small molecules to pass the plasma membrane, including release of ATP into the extracellular space.23,36,37 A possible role for ATP in MSC differentiation is supported by the observation that ATP hydrolysis reduces heterotopic ossification after injury. 38 Notably, EF-stimulated MSC also released ATP to the extracellular space; furthermore, extracellular ATP supplement in the absence of EF immediately simulated the peculiar pattern of calcium activation shown under EF stimulation. This finding suggested that ATP, possibly released from Cx43 hemichannels, may trigger L-type calcium channel activation (Fig. 6). The EF might drive molecules such as ATP through Cx43 hemichannels and gap junctions, leading to increased Ca2+ influx through L-type Ca2+ channels in an ATP-dependent manner.
The physiological importance of Cx43 was demonstrated in human Cx43 gene mutations and Cx43 ablated mice showing skeletal malformations.17,39 Regarding our results showing that connexin 43 and gap junctions were required for the EF-induced calcium rise and synchronized osteogenic gene expression, Cx43 might be an important target for EF-driven bone tissue engineering. Since Cx43 hemichannels are found in several cell types other than bone cells, such as cardiomyocytes, 15 MDCK, HeLa cells, 40 basal keratinocytes, 41 and astrocytes, 42 the contribution of Cx43 hemichannels to EF-triggered migration and differentiation in other cell types may be further elucidated.
In addition, our results show that simultaneous calcium activation in MSC under EF seems to be largely dependent on EF-induced gap junctional activation. MSC could propagate [Ca2+]i increase into neighboring cells in an efficient and orderly way, like a calcium wave, by EF-induced gap junctional activation. We frequently observed a regular sequence of the propagation: following a latency of about 30 − 40 min, a single MSC elevated [Ca2+]i; after 10 − 20 min, neighboring cells and subsequently cells in the vicinity are activated. These findings implicate that in vivo a small number of EF-triggered MSC may efficiently transmit [Ca2+]i toward neighboring cells for synchronized osteogenic induction. Our data show that the EF-triggered [Ca2+]i rise further activates downstream signaling, including calmodulin/calcineurin, inducing osteogenic gene expressions possibly via NFAT/osterix (Fig. 6).
Conclusions
In summary, on biocompatible implant surfaces such as TiO2 nanotubes, MSC sense and react to both x-y planar and z-axis-directed EFs by increased intracellular Ca2+ levels, resulting in osteogenic induction. Electric stimuli induce membrane protrusions with Cx43 transport to plasma membrane, and Cx43 seems to be a key EF-sensing mediator for intracellular calcium elevation and osteogenic differentiation of MSC. Our x-y planar and Z-axis-directed EF application system may be further applied on 3D-biocompatible substrates in bone tissue engineering. The identification of Cx43-mediated EF stimulation in osteogenic differentiation may contribute to the development of new therapeutic interventions on bone repair and tissue engineering.
Footnotes
Acknowledgments
We thank Sandra Lehnert for providing a plasma membrane-targeted EGFP plasmid. Dr. Sebastian Bauer is acknowledged for early preliminary work. This work was supported by DFG grant PA 2537/1-1 to J.P. and P.S.
Author Contributions
J.P. and A.M. designed and conducted the experiments. A.M. generated nanostructured materials. A.M. and J.P. performed electron microscopy. J.P. and M.F. performed cellular experiments, data and statistical analysis. H.S., K.vdM., and P.S. interpreted the data. J.P., A.M., and M.F. wrote the article with substantial inputs from H.S., K.vdM., and P.S.
Disclosure Statement
All authors state that they have no conflicts of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.