
Abstract
Ewing's sarcoma (ES) is a poorly differentiated pediatric tumor of aggressive behavior characterized by propensity to metastasize to bone. Interactions between the tumor and bone cells orchestrate a vicious cycle in which tumor cells induce osteoclast differentiation and activation to cause osteolytic lesions, broken bones, pain, and hypercalcemia. The lack of controllable models that can recapitulate osteolysis in ES impedes the development of new therapies and limits our understanding of how tumor cells invade bone. In response to this need, tissue-engineered models are now being developed to enable quantitative, predictive studies of human tumors. In this study, we report a novel bioengineered model of ES that incorporates the osteolytic process. Our strategy is based on engineering human bone containing both osteoclasts and osteoblasts within three-dimensional mineralized bone matrix. We show that the bone matrix is resorbed by mature osteoclasts while the new bone matrix is formed by osteoblasts, leading to calcium release and bone remodeling. Introduction of ES cell aggregates into the bone niche induced decreases in bone density, connectivity, and matrix deposition. Additionally, therapeutic reagents, such as zoledronic acid, which have demonstrated efficacy in ES treatment, inhibited bone resorption mediated by osteoclasts in the tumor model.
Introduction
H
Under physiological conditions, bone is remodeled in a fine-tuned process by which osteoblasts produce new extracellular matrix of the bone and osteoclasts resorb old bone. During this process, minerals (i.e., calcium and phosphorus), growth factors, and cytokines are released from the bone matrix to maintain mineral homeostasis and acid–base balance in the body. 6 However, the crosstalk between tumor cells, osteoblasts, and osteoclasts disrupts the bone remodeling and initiates either bone destruction (osteolytic tumors) or abnormal bone formation (osteoblastic tumors). 7
Ewing's sarcoma (ES) is the second most frequent bone tumor affecting children and young adults that arises and metastasizes in bone. 8 It is characterized by fast growth and progressive bone destruction by osteolysis. 9 Notably, ES cells are incapable of directly degrading bone matrix. 10 Instead, they orchestrate the process of bone resorption through a vicious cycle of recruitment and activation of osteoclasts that is mediated by osteoblasts.11,12 Bone destruction by osteoclasts releases calcium and growth factors from the bone matrix that favor acidosis and tumor growth and thereby the activation of osteoclasts and increased bone resorption. 13
The lack of ability to replicate in vitro the bone osteolysis associated with the ES represents a critical barrier to understanding of the mechanisms underlying tumor progression and evaluating new therapeutics. Bioengineered tumor models are becoming invaluable tools for cancer research.14,15 However, modeling the bone invasion by cancer remains a challenge. Due to the intrinsic biology of osteolytic tumors, it is of paramount importance to include both osteoblasts and osteoclasts into the bone that will be populated by cancer cells, within the mineralized bone matrix.
In this study, we describe a complex bioengineered model of ES tumor within the human bone niche, recapitulating the osteolytic process observed in patients. To this end, we have engineered a living bone niche comprising osteoclasts and osteoblasts that provides a controlled biomimetic environment for ES growth (Fig. 1). To illustrate the utility of this model, we conducted testing of antiosteolytic drugs.
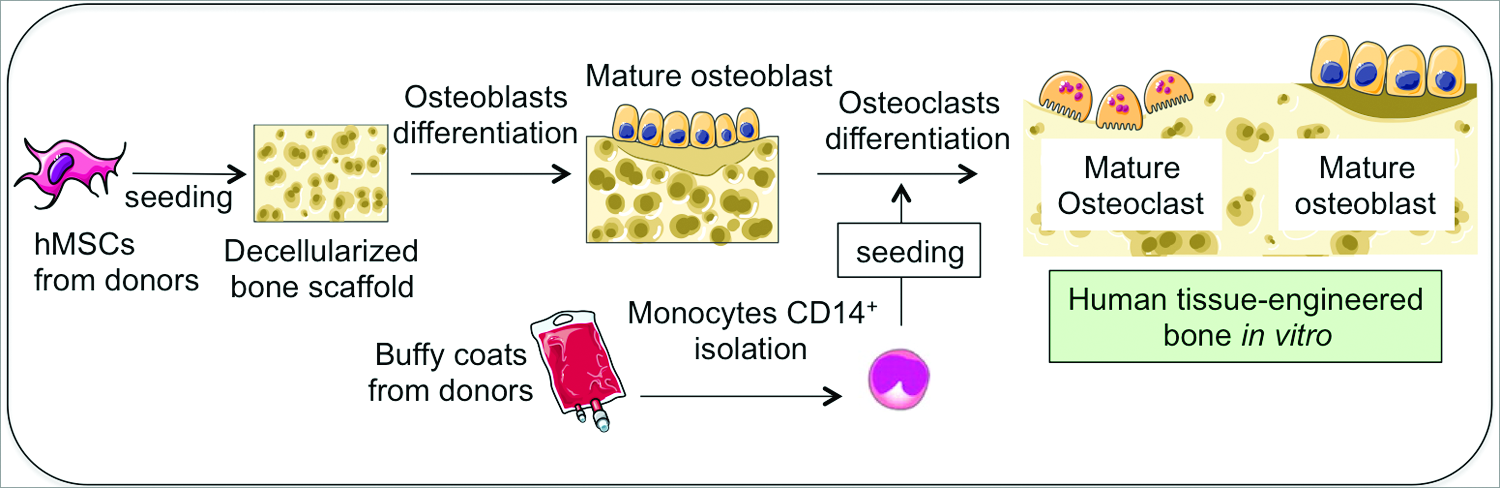
Overall approach to build a human tissue-engineered bone in vitro containing osteoblasts and osteoclasts. hMSC isolated from bone marrow aspirates are seeded within a decellularized bovine bone scaffold and differentiated toward mature osteoblasts. Then, monocytes CD14+ isolated from buffy coats from human blood are cocultured with osteoblasts and differentiated into mature osteoclasts. hMSC, human mesenchymal stem cells. Color images available online at
Results
Derivation of bone cell precursors
Since the first study describing the capability of human mesenchymal stem cells (hMSC) to differentiate into osteoblasts, 16 hMSCs from various sources have been used to engineer bone. Our laboratory has long-standing interest in engineering human bone by cultivation of hMSC in decellularized bone scaffolds, using bioreactors with medium perfusion.17–20 The decellularized bone scaffold preserves not only the structural and mechanical features of the original bone, but also maintains its inorganic mineral phase and many of the growth factors. 21 Notably, owing to the highly osteogenic properties of these scaffolds, the supplementation of BMP-2 during bone tissue engineering is not necessary.22,23 When studied in orthotopic large animal models of bone repair, bone engineered using this approach provided mechanical support and integrated with the surrounding tissues, with active tissue remodeling. 23
Following our well-established protocol, we used hMSC as a source of osteoblasts for engineering bone in vitro. First, we confirmed the ability of hMSC (from two different donors) to differentiate into osteoblasts, both in cell monolayers and in decellularized bone scaffolds (Supplementary Figs. S1A and S2A; Supplementary Data are available online at
We then assessed the capability of osteoclast precursors (CD14+ monocytes) to differentiate into mature osteoclasts24,25 (Supplementary Fig. S3A) that were identified based on their unique morphology and function. Osteoclasts are large, multinucleated, and polarized cells with the nuclei localized toward the apical membrane and a ruffled border membrane. These cells are specialized in bone resorption that proceeds with degradation of organic matrix and demineralization of the mineral matrix in specific regions known as “resorption lacunae,” and inducing increases in local concentrations of calcium and phosphate. Activated osteoclasts resorb bone by lowering the pH in the resorption lacunae, following secretion of acidic hydrolases such as cathepsin K and the tartrate-resistant acid phosphatase (TRAP), and express considerable levels of calcitonin receptor.26,27
Osteoclasts were derived from human monocytes isolated from buffy coats, and tested for purity. On average, the enrichment of CD14+ monocytes was 94%, as determined by flow cytometry analysis (Supplementary Fig. S3B). The purified monocytes were cultured for up to 3 weeks in monolayer in the presence of RANKL to induce osteoclastic lineage differentiation. 24 By week 1, the osteoclast markers, TRAP, calcitonin receptor, and cathepsin K, were expressed (Supplementary Fig S3C), and this expression reached the maximum level at week 3. We evaluated the morphology, differentiation, and multinuclearity of osteoclasts by TRAP staining (Supplementary Fig S3D). Osteoclast activation and functionality (Supplementary Fig. S3E), and the calcium release over time were evaluated and compared with the undifferentiated cell control (Supplementary Fig. S3F).
Engineered human bone containing osteoblasts and osteoclasts
The methods used to engineer human bone containing both osteoblast and osteoclast cell populations were based on the previous model of engineered bone containing only osteoblasts (Supplementary Fig. S2). Initial experiments were done to set up a suitable protocol for coculturing both types of bone cells in the three-dimensional environment. It is important that the average lifespan of human osteoclasts is only 2–4 weeks, whereas the average lifespan of osteoblasts is as long as 3 months.28,29
Thus, first we differentiated hMSC into osteoblasts within a decellularized bone scaffold for 3 weeks (TE-hOB). Then, we introduced the CD14+ monocytes into the TE-hOB and cultured them in an osteoclastogenic differentiation medium for 1–3 weeks, to generate 3D bone with osteoclasts (TE-hOB + hOC; Fig. 1). Initially (at week 1) we could not identify morphological differences between the TE-hOB with and without monocytes by histological analysis (H/E staining; Fig. 2A and Supplementary S4A). At weeks 2 and 3, osteoclasts were clearly identified in the construct by morphological analysis (Fig. 2A and Supplementary S4A, B). Resorption lacunae were visible in the TE-hOB + hOC (Fig. 2A). In coculture with osteoclasts, osteoblasts retained their characteristic hexagonal morphology 30 and formed compact tissue layers by week 2 (Supplementary Fig. S4C). These results confirmed feasibility of generating in vitro an engineered bone niche comprised of both osteoclasts and osteoblasts.

Characterization of osteoclasts within a tissue-engineered bone containing osteoblasts.
To evaluate osteoclast differentiation and activity, we analyzed the level of mRNA expression of calcitonin receptor and TRAP, and the genes expressed in osteoclasts and involved in cell differentiation 31 in both types of constructs (hOB; hOB + hOC). Consistent with morphological studies, we observed an increase in calcitonin receptor and TRAP expression at week 2 (Fig. 2B). As expected, and consistent with the lifespan of human osteoclasts, calcitonin receptor (CT-R) mRNA levels showed a trend of decrease at week 3 (Fig. 2B). In parallel with the increasing expression of osteoclast markers, we observed significantly decreased levels of the monocyte marker CD14 at mRNA levels at week 2 and 3 (Fig. 2C).
We assessed osteoclast activation by TRAP staining and calcium release to confirm the presence of TRAP+ cells at week 2 (Fig. 2D). The highest levels of Ca2+ release activity were recorded at week 2 (Fig. 2E), suggesting 2 weeks as the most suitable time point for osteoclast studies in the 3D construct. The mRNA levels of expression of osteoblast markers, OPN and BSP, 31 increased by week 2, as determined by qRT-PCR (Fig. 2F). OPN expression was not restricted to osteoblasts, as both osteoblasts and osteoclasts are capable of synthesizing OPN. 32 These data confirm the presence of osteoclasts in coculture with osteoblasts in the bone tissue construct.
Then, we studied the noncellular bone compartment by assessing the bone microstructure with and without osteoclasts by microcomputed tomography (μCT) scans, to obtain quantitative bone structural parameters (Fig. 3A). We observed no differences in bone volume density (BV/TV) between the groups, suggesting a balance between bone production and bone resorption. A number of studies have demonstrated a direct relationship between osteoclast activity and bone remodeling and an increase in connectivity density.33,34 Importantly, and as expected, the connectivity density (Conn D) was slightly higher in the group with osteoclasts (Fig. 3B). These data suggest that osteoclasts are metabolically active and capable of resorbing bone.

Evaluation of bone microstructure in the TE bone.
We also analyzed the nonmineralized extracellular matrix component of the bone. We focused on BSP, as an important marker related to bone turnover that can be detected in serum, which also enhances osteoclast-mediated bone resorption. 31 Interestingly, we found differences in BSP protein distribution between both groups by immunohistochemistry (Fig. 3C). The group with only osteoblasts exhibited uniform distribution of BSP, in contrast to mosaic-patterned patches of BSP in the group with osteoclasts, consistent with bone remodeling in vitro.
We then investigated the effects of a therapeutic reagent zoledronic acid (ZA) that has demonstrated efficacy in patients. ZA is a bisphosphonate commonly used to treat osteoclast-mediated bone loss in people with osteoporosis. The mechanism of action of ZA consists of inducing apoptosis in osteoclasts and inhibiting osteoclast-mediated bone resorption. 35 To determine whether osteoblasts are still capable of producing matrix after coculture with osteoclasts, we inhibited osteoclasts with ZA (20 μM for 2 days), and evaluated BSP distribution by immunohistochemistry (Fig. 3C). We observed a partial recovery of uniform BSP distribution in tissue constructs and, quite surprisingly, large patches of strong BSP staining after ZA treatment. This result confirms that osteoblasts are active and producing new matrix, which recapitulates in vitro the drug function observed in clinic. The response of tissue-engineered (TE) bone to ZA treatment was similar to those observed in animal models 36 and patients, 35 reinforcing the biomimetic value of the bone-engineered constructs comprising both osteoblasts and osteoclasts.
TE model of ES
To build the tumor model, we generated a TE bone containing mature osteoblasts and mature osteoclasts, differentiated for 12 days. We then infused ES aggregates (cultured for 1 week to allow aggregate formation) into the construct and cultured the tumor model for an additional 1 week (Fig. 1). Two different ES models were generated: type 1 (using SK-N-MC cell line) and type 2 (using RD-ES cell line). We confirmed the presence of cancer cells in the tumor model by morphological studies (Fig. 4A) and by evaluating the expression levels of EWS/FLI and NKX2.2 genes that are specifically expressed at high levels in ES (Fig. 4B). Additionally, we observed decreases in BSP levels in the tumor model relatively to the corresponding bone constructs (Fig. 4C).

Generation and characterization of the TE model of ES.
Interactions between cancer cells and bone cells orchestrate a vicious cycle in which tumor cells induce osteoclast activation and osteoblast inhibition, resulting in bone resorption and osteolysis. 11 To evaluate the possible effect of ES cells on bone resorption, we performed μCT scans of the tumor models with RD-ES and SK-N-MC cells (Fig. 5A). Consistent with the previous studies in animal models37,38 of bone osteolytic tumors,33,34 we observed a marked decrease in the trabecular number in the constructs with ES cells (Fig 5.B). Although not significant, the same tendency was observed for the Conn D parameter (Supplementary Fig. S5). Conversely, an increase for trabecular space values was found (Fig 5B). These results suggest that Ewing's sarcoma cells induce bone resorption and osteoclast activation. We did not observe any differences in bone volume density per unit tissue volume (BV/TV) and trabecular thickness (Supplementary Fig. S5). We quantified calcium release in tumor model supernatants, but did not observe any difference compared with the bone construct with osteoclasts. Together, these data suggest that the TE model of ES is mimicking, at least in part, the first steps of the osteolytic process.
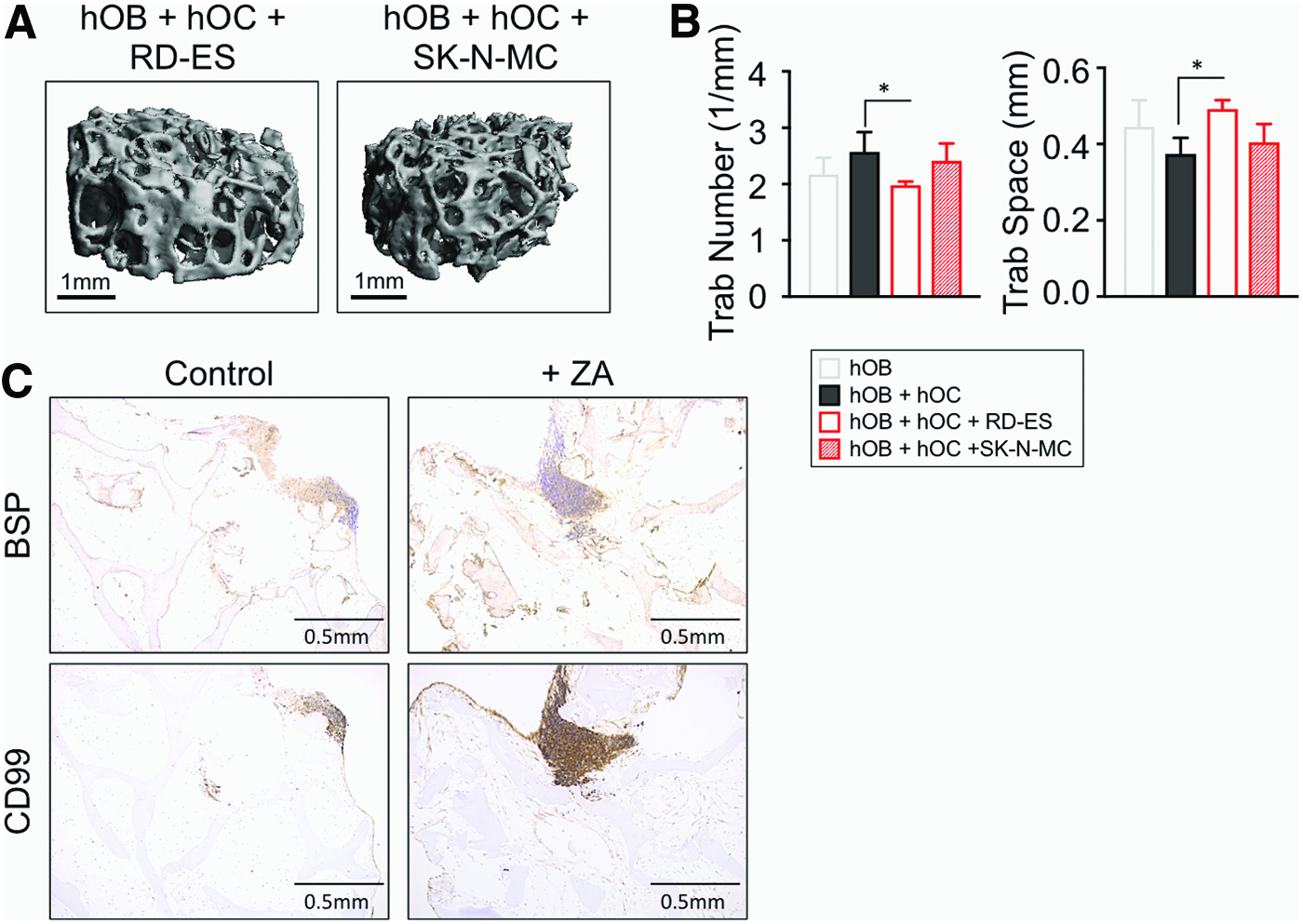
Analysis of bone microstructure and zoledronic acid effects in the TE model of ES.
For further characterization, we examined BSP distribution in the tumor model by immunohistochemistry. As expected, bone extracellular matrix lacked BSP, but surprisingly, we observed BSP colocalized with the CD99 ES marker (Fig. 5C). We recently reported the capability of ES cells to produce BSP in a 3D environment. 39 Thus, BSP observed in Figure 5C could be secreted by ES cells, and not by the osteoblasts. This result reinforces the idea of ES cell-mediated bone matrix degradation.
ZA has been shown to target both osteoclasts and ES cells. 40 To determine whether cancer cells inhibit the ability of osteoblasts to produce BSP, we treated the tumor model with ZA (20 μM for 2 days). BSP was detectable in the whole construct after treatment that suggests reactivation of osteoblasts (Fig. 5C), recapitulating the effects observed in mice models. 40
Discussion
In the last 20 years, a number of two-dimensional (2D) cultures and animal models of ES have contributed critical information about cancer biology and served as preclinical systems for therapeutic screens. 4 Unfortunately, the existing ES models have failed to faithfully predict human physiology and support the development of effective treatment modalities. In spite of large investments, the use of these models have delayed drug discovery 41 and exposed children to unnecessary chemicals, suggesting that modeling of the tumor progression requires interactions between tumor cells and their surrounding microenvironment. 42
Recently, TE models have started to bridge the gap between 2D in vitro cultures (used for discovery and screening) and in vivo animal models (used for efficacy and safety assessment before proceeding to clinical trials) providing a predictive, inexpensive, and low time-consuming alternative. However, recapitulating tumor features in vitro is still a major challenge in the field. 15 In this study, we address this challenge by developing a novel protocol for engineering human bone comprising the key components that are crucial for the vicious cycle of cancer to bone: osteoclasts, osteoblasts, bone ECM secreted by cells, native mineralized ECM, and ES cells. To this end, the healthy bone tissue was engineered by coculturing osteoblasts (derived from hMSC) and osteoclasts (derived from monocytes isolated from blood samples).
We have previously demonstrated17–19 that at least 3 weeks of cultivation is necessary for hMSC to differentiate into osteoblasts. In this study, we used again this robust differentiation protocol and showed expression of high levels of osteoblast markers. Next, we cocultured CD14+ monocytes with the bone engineered from osteoblasts only for 1, 2, or 3 weeks in an osteoclastogenic differentiation medium. We clearly identified osteoclasts at week 2 by morphological analysis, expression of osteoclasts markers, and activity assays that confirmed physiological bone remodeling in vitro. Because the average lifespan of human osteoclasts is about 2–4 weeks,28,29 it is not surprising that at week 2 we found the maximum peak of activity and after that, we observe a slight decrease in all the readouts.
After 12 days of osteoclast differentiation, we infused ES aggregates into the TE bone, and maintained this tumor model for 1 week to secure the activity of the osteoclasts. We observed that the living TE bone niche provided a biomimetic and controlled environment for recapitulating ES growth and development. ES cells cultured in this niche recapitulated the initial steps of lytic lesions found in the patient's tumors (i.e., loss of BSP, decrease in trabecular number, and increase in trabecular space, as well as a slightly decreased Bone Volume Density and Conn D). It would also be interesting to model further steps of the osteolytic process. Additionally, it had effects in the TE model that was comparable to those observed in animal studies.
ZA is a common bisphosphonate used in clinic, and its effects on inhibition of osteoclasts have been extensively studied.43,44 Although no negative effect on osteoblasts has been reported yet, little is known about the role of ZA in regulating osteoblast functions. Interestingly, recent reports showed that ZA could induce BSP expression directly in osteoblast-like cells through inactivation of Rho GTPases45,46 Although it was beyond the scope of this study, our model would also be useful for further characterization of the mechanism of action of ZA in osteoblasts and for studies of osteoporosis.
An obvious goal of cancer engineers is to develop better protocols and technologies for generating in vitro human tumor models predictive of native tumors. 47 Thus, a desirable objective is to establish a personalized approach to studies of osteolysis in ES using the patient's own cells. Recently, a method for deriving macrophages from iPS cells has been established. 48 Notably, macrophages are capable of differentiating into osteoclasts. Additionally, we reported a protocol to engineer osteoblast-based bone using iPS cells. It would be interesting to use the same iPS cells from an ES patient to form a personalized bone niche. Macrophages are known to have a role in cancer progression and bone remodeling. 49 Our model would be an excellent system to evaluate macrophage interactions with bone cells and cancer cells as well. Also, these analyses would give us the opportunity to further characterize and to better understand cell fate of those monocytes that potentially do not differentiate into osteoclasts.
TE models of human tumors are now designed to conform to the three R's: Reduction, Refinement, and Replacement. This new generation of models of ES can faithfully recapitulate the osteolytic process observed in patients' bones. While animal models have limitations, they display a range of complexity associated with systemic factors that TE systems still lack. A challenging and desirable goal would be to engineer a bone niche that can maintain osteoclast and osteoblast precursors in undifferentiated state, to maintain active osteolysis, and self-renewal over long periods of time. A less biomimetic, but perhaps more feasible option would be to introduce medium perfusion into the system, and to infuse bone precursors at timed intervals. We believe that the field of cancer engineering is moving fast, and in a good direction. The proposed model has high transformative potential, as it enables critical advances in tumor modeling under conditions predictive of human physiology.
Materials and Methods
Cell culture
Cancer cell lines
ES cell lines SK-N-MC (HTB-10) and RD-ES (HTB-166) were purchased from the American Type Culture Collection (ATCC) and cultured according to the manufacturer's specifications. RD-ES cells were cultured in ATCC-formulated RPMI-1640 Medium (RPMI) and SK-N-MC cells were cultured in ATCC-formulated Eagle's Minimum Essential Medium (EMEM). Both media were supplemented with 10% (v/v) HyClone fetal bovine serum (FBS) and 1% penicillin/streptomycin. Cells were cultured at 37°C in a humidified incubator at 5% CO2.
Tumor aggregates
To generate tumor aggregates, aliquots of 300,000 ES cells were centrifuged in 15-mL Falcon tubes (5 min at 12,000 rpm), and cultured in 4 mL of osteoclast differentiation medium without cytokines: Minimum Essential Medium Eagle Alpha modification, consisting of α-MEM (M4526; Sigma) supplemented with 10% (v/v) heat-inactivated HyClone FBS, 1% penicillin/streptomycin, and
Human mesenchymal stem cells
Unprocessed human bone marrow aspirates were purchased from Lonza. We used aspirates from two different donors: donor 1 (code 26737) and donor 2 (code 26798). hMSCs were isolated from these aspirates, characterized, and prepared as in our previous studies.22,23,50
Derivation of osteoblasts from hMSCs
Cell culture and differentiation into osteoblasts were carried out as in our previous studies. 48 Briefly, hMSCs were cultured in expansion medium (DMEM supplemented with 10% (v/v) HyClone FBS, 1% penicillin/streptomycin, and 1 ng/mL of basic fibroblast growth factor b, bFGF). Differentiation into osteoblasts was performed by culturing hMSC in osteoblast differentiation medium (DMEM supplemented with 10% v/v HyClone FBS and 1% penicillin/streptomycin, 1 μM dexamethasone, 10 mM β-glycerophosphate, and 50 μM ascorbic acid-2-phosphate) for 3 weeks. hMSC and osteoblasts were cultured at 37°C in a humidified incubator at 5% CO2.
Isolation of monocytes
Peripheral blood mononuclear cells (PBMC) were isolated from buffy coats of human blood (fully deidentified samples obtained from the New York Blood Center) by density gradient centrifugation with Ficoll-Paque PLUS (17-1440-02; GE Healthcare). Monocytes were derived from the PBMC preparations by immunomagnetic isolation (The Big Easy EasySep Magnet, #180001; Stem Cell Technologies) using negative selection (the EasySep Human Monocyte Isolation Kit #19359; Stem Cell Technologies), following the manufacturer's protocol. Then, 8 × 106 monocytes were cultured on 25-cm2 ultralow attachment flasks (#3815; Corning) with 10 mL of maintenance medium: RPMI 1640 (30-2001; ATCC) supplemented with 10% heat-inactivated human serum (#35-060; Corning), 1% penicillin/streptomycin, 20 ng/mL Recombinant Human M-CSF (#300-25; PeproTech) during 2 days at 37°C in a humidified incubator at 5% CO2.
Derivation of osteoclasts from human monocytes
Human CD14+ monocytes were incubated with a differentiation medium consisting of Minimum Essential Medium Eagle Alpha modification (α-MEM, M4526; Sigma) supplemented with 10% (v/v) heat-inactivated HyClone FBS, 1% penicillin/streptomycin,
Resorption pit assay
Human CD14+ monocytes were plated into 24-well osteo assay plate (100,000 cells/well) (#3987; Corning) and cultured either in a complete osteoclast differentiation medium, or without sRANKL as a control for cell differentiation. At different time points, 10% bleach solution was added to each well and cells were incubated for 10 min at room temperature. Then, wells were washed three times with distilled water and air dried overnight. Resorption pits were visualized at 10 × magnification and, for improving the quality of the image, a blue filter was used.
Engineered bone tissue containing osteoblasts and osteoclasts
Scaffolds (4 mm diameter × 4 mm high plugs) were prepared from decellularized bovine bone as in our previous studies. 50 hMSC (1.5 × 106 per scaffold) were seeded into each scaffold and cultured with osteoblast differentiation medium for 3 weeks, with a complete medium change twice a week.
Bone constructs were then incubated in osteoclast differentiation medium without cytokines (M-CSF and sRANK Ligand) for 1 h, and bisected. One half of the tissue construct was placed into a 4 mm × 4 mm (inner diameter × height) PDMS ring and cultured with the addition of 500,000 osteoclasts in 10 μL of osteoclast differentiation medium for 30 min at 37°C in a humidified incubator at 5% CO2. The construct was flipped and seeded again with 500,000 osteoclasts in 10 μL of osteoclast differentiation medium for 30 min at 37°C in a humidified incubator at 5% CO2.
The resulting constructs were placed into low-attachment six-well plates (1 construct/well) containing 5 mL of osteoclast differentiation medium. Medium was changed twice a week. This group was termed hOB + hOC. The other half of each tissue construct that contained only osteoblasts was termed the hOB group, and cultured with osteoclast differentiation medium without cytokines.
Tissue-engineered tumor model
Tumor cells were introduced into the osteoblast–osteoclast bone niche using methods from our previous studies. 51 Aggregates of ES cells (RD-ES or SK-N-MC cell lines) containing 0.3 × 106 cells were injected into the tissue constructs (3 aggregates/construct) and the resulting cancer cell–bone constructs were cultured for 1 week in an osteoclast differentiation medium without supplemental cytokines. This group was termed hOB + hOC + RD-ES or hOB + hOC + SK-N-MC, depending on the ES cell line used for model generation. Bone tissue constructs (hOB + hOC) without cancer cells were used as a control. Constructs were treated with 20 μM of ZA (#SML0223; Sigma-Aldrich for 2 days)
Quantitative real-time PCR
The qRT-PCR was carried out using DNA Master SYBR Green I mix (Applied Biosystems), and as we previously described
51
mRNA expression levels were quantified applying the ΔCt method, ΔCt = (Ct of gene of interest − Ct of GAPDH). qRT-PCR primer sequences that were obtained from the PrimerBank database (
The sequences were obtained from the PrimerBank Database (
Histology and immunohistochemistry
Tumor tissue constructs and all controls were fixed in 10% formalin for 24 h and then decalcified with Immunocal (StatLab Corp., McKinney, TX) for 2 days. Samples were dehydrated in graded ethanol washes, and embedded in paraffin. Serial sections (3 μm thick) were prepared for histology and stained with Hematoxylin and Eosin (H&E).
Immunohistochemistry stainings were performed using primary antibodies specific to CD99 (dilution 1:500; SIG-3620; Signet antibodies) and bone sialoprotein (dilution 1:500, ab33022; Abcam), and developed using the Vector Elite ABC Kit (Vector Laboratories), following the manufacturer's instructions. Briefly, sections were blocked with serum for 30 min and incubated with the primary antibody overnight at 4°C. After washing with PBS, samples were incubated with secondary antibodies and developed (Vector Laboratories). Negative controls were prepared by omitting the primary antibody step. Alkaline phosphatase and von Kossa stainings were performed as previously described. 48 TRAP staining was performed using the K-assay (#KY-008; Kamiya Biomedical Company).
Monocytes (300,000/well in six-well plates) were cultured in a complete osteoclast differentiation medium, or without sRANKL as a control for differentiation. At timed intervals (1–3 weeks), culture medium was removed and cells were fixed and stained for TRAP, by following the manufacturer's protocol.
TE bone constructs were fixed in 10% formalin, decalcified in 12.5% EDTA, embedded in paraffin, sectioned to 4 μm, stained for TRAP according to the manufacturer's instructions, and counterstained with Hematoxylin QS (Vector Labs).
Scanning electron microscopy
Scanning electron microscopy (SEM) samples were prepared by fixing in 4% paraformaldehyde solution for 1 h. After rinsing twice with PBS, samples were subsequently dehydrated in increasingly concentrated aqueous solutions of ethanol (70%, 85%, 95%, and 100%) for 5 min each, and finally hexamethyldisilazane (HMDS) for 15 min. After air drying overnight in a chemical hood, the samples were sputter coated with ∼10 nm of gold and palladium. SEM images were acquired using a Hitachi S-4700 FE-SEM.
Calcium release analysis
Supernatants of culture medium were sampled (1 mL/sample), snap frozen in liquid nitrogen, and stored at −80°C. The Ca2+ concentrations were analyzed using the Ca2+ Detection Kit (Abcam, ab102505) following the manufacturer's protocol. Briefly, supernatants were centrifuged for 2–5 min at 4°C at top speed using a cold microcentrifuge to remove any insoluble material. Supernatants were collected and transferred to clean tubes. Ninety microliters of the chromogenic reagent were added to each sample. The chromogenic complex formed between calcium ions and 0-cresolphthalein was measured using a microplate reader at OD = 575 nm. The measured absorbance values for each standard were plotted as a function of the final concentration of calcium. Finally, the calcium concentrations in the samples were calculated from the standard curve.
Microcomputed tomography
Samples were scanned and analyzed using a Scanco VivaCT 40 microcomputed tomography system (Scanco Medical, Basserdorf, Switzerland). Scans were performed using 55 kVp, 109 μA, and 200 ms integration time, and resulted in images with 21 μm isotropic voxel size. Reconstructed images were smoothed using a Gaussian filter (sigma 0.8, support 1), segmented using a global threshold of 30% maximum gray-scale value, and processed using the standard trabecular morphometry evaluation.
Author Contributions
Dr. Villasante conceived the project, developed the experimental design, performed most of the experiments, analyzed data, and wrote the article. Mr. Marturano-Kruik performed calcium assays and histological and immunohistochemical stainings. Mr. S.T. Robinson and Dr. X.E. Guo conducted μCT studies and analyzed data. Ms. Liu performed SEM experiments. Dr. Vunjak-Novakovic supervised the study, contributed to data analysis and interpretation, and wrote the article.
Footnotes
Acknowledgments
The authors gratefully acknowledge NIH funding of this work (grants EB002520 and EB17103 to G.V.-N.). They also acknowledge expert help of Dr. Llucia-Valldeperas and Dr. Freytes (isolation of monocytes), Histology Facility at Columbia University (histological services) and Dr. Ho, Flow Cytometry Core Facility at the Columbia Center for Translational Immunology (cell sorting).
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.