
Abstract
The GID SVF-1 and GID SVF-2 are closed-system, disposable, scalable cellular isolation devices designed for isolating the human adipose-derived stromal vascular fraction (hSVF) from lipoaspirates at the clinical point of care. The purpose of this study was to characterize the device performance with respect to cell yield and viability of the hSVF, as well as the hSVF purity and cellular composition. Our results demonstrate that adipose-derived hSVF can be safely obtained using both devices and standardized methods, yielding cells that are free of bacterial contaminants as measured by endotoxin levels, Gram stain, and culture. In addition, they produce cellular preparations with compositions consistent with reported values within the peer-reviewed literature using similar methods.
Introduction
A
Currently, there are several devices in various stages of development for the isolation of SVF cells at the PoC. Although few direct comparisons of these different devices exist, it is clear that each is unique—differing in matters of complexity, automation, cost, efficiency, and efficacy. Nevertheless, each will be held to the same regulatory requirements and quality control and assurance expectations. That is, any device or system and associated protocol that intend to translate adipose-derived stromal vascular cell therapies to the clinic must demonstrate dependable methods that yield consistent “cell-product” output characterized at least in terms of identity, purity, and potency. The ideal cell isolation device would be simple to understand and use, affordable and efficient, scalable and flexible, and consistently produce a cell output that is biologically viable and active. The purpose of this study was to characterize the identity, purity, and reproducibility of hSVF cells isolated using a novel, scalable, closed-system, disposable, device platform at the PoC, and to compare findings to those reported in the literature for similar platforms.
Materials and Methods
Adipose tissue was obtained from elective liposuction surgeries under an IRB-approved protocol at the University of Florida, Gainesville.
Tissue harvest and SVF isolation
Abdominal adipose tissue was harvested using machine-based, tumescent liposuction. Aspirated tissue was collected into a sterile device (GID SVF-1 or GID SVF-2) connected directly in-line to the aspiration cannula and suction tubing (Figs. 1 and 2). The GID SVF-1 and GID SVF-2 devices are packaged and sold sterilely according to industry standards, and differ only in the maximum volume of adipose tissue (not lipoaspirate) that can be processed in a single run (for SVF-1, up to 300 mL; for SVF-2, up to 125 mL). Sterility is achieved via E-beam radiation. The devices allow selective capture of tissue fragments in an inner mesh filter compartment, while waste fluid readily passes through and is immediately aspirated into a waste container. After lipoharvest was complete, the retained tissue in the device was rinsed with Ringer's lactate solution (LR). The GID SVF device was then filled with warmed 37°C LR and insulated with heating packs for transportation to the laboratory.

Device design.

Outline of device function. Unprocessed lipoaspirate (1) is captured within the internal mesh “trap” and rinsed several times for tissue “cleaning” (2) before enzymatic dissociation in a warmer-shaker. After enzymatic incubation, the device is then placed in a tabletop centrifuge and the SVF cells are concentrated into a pellet (3). Finally, the cell pellet is removed by sterile aspiration by needle perforation of the internal mesh via the central port (4, 5). 5a, 14G spinal needle; 5b, central port; 5c, collagenase-digested sample; 5d, internal mesh; 5e, sump-tube vent; 5f, SVF cell pellet. Color images available online at
On arrival in the laboratory, tissue processing occurred within a laminar flow hood even though the device enables a closed-system processing method from start to finish. The tissue was washed with warm solution containing 20 μg/mL of ciprofloxacin and 5000 U/L of injectable heparin until the effluent was clear, leaving “dry” and uniformly yellow adipose tissue inside the mesh compartment (Fig. 2).
The canister was weighed, and tissue mass and volume determined for further processing. An equal volume of 37°C LR was then added to the canister to create a “total catalytic volume” (TCV). The adipose was disaggregated using type I collagenase CLS-1 (Worthington, Lakewood, NJ) at 200 CDU/mL of the TCV. The collagenase was injected into the canister through a sterile 0.22-μm filter (Millex-MP, Millipore, Cork, Ireland). The device with adipose, buffer, and collagenase was then placed into a 38°C incubated shaker for 40 min at 150 rpm. After disaggregation, human albumin solution was added to achieve a concentration of 2.5% to reduce collagenase activity, and the lipid-laden, mature adipocytes were subsequently separated from the remaining cellular and liquid fractions using centrifugation. The device was centrifuged at 800 g for 10 min using a standard laboratory centrifuge (Sorvall ST40; Thermo Fisher Scientific, MA) and rotor (BIOLiner swinging-bucket; Thermo Fisher Scientific, MA). The SVF-2 protocol was slightly modified in respect to the centrifugation secondary to difference in lipoaspirate sample size. The SVF-2 device was centrifuged at 600 × 3 g for 6 min, stirred with the internal impeller, and then centrifuged for another 4 min for a total of 10 min of centrifugation time. After centrifugation of the dissociated cell suspension, the resulting floating layer (i.e., buoyant layer of mature adipocytes and free oil) was easily removed via aspiration through a device port, leaving an aqueous layer and the SVF cell pellet. The cell pellet was accessed through the device's central port with a 14G spinal needle and suspended in sterile LR (10 mL for SVF-1 cells; 4 mL for SVF-2 cells) (Fig. 2).
SVF counting and viability
A 0.5 mL sample of cell suspension was placed into a 1.5-mL Eppendorf tube for cell counting and assay. Cells were counted using a NucleoCounter (NC-100; New Brunswick Scientific) according to the manufacturer's instructions. For erythrocyte lysis, the remaining SVF suspension was centrifuged at 400 g for 5 min and suspended in ammonium–chloride–potassium (ACK) lysing buffer (KD Medical RGF-3015) and dark incubated at ambient room temperature (∼21°C) for 5 min. The suspension was then passed through a 100-μm nylon mesh filter (Millipore SCNY00100). After another centrifugation and disposal of the supernatant, the cell pellet was resuspended and an aliquot taken for an additional cell count (post-red blood cell [RBC] lysis and filter).
Residual collagenase
Collagenase levels were quantified throughout the isolation process and at the completion of the process by using the EnzChek Gelatinase/Collagenase Assay Kit according to the manufacturer's instructions (Cat No. E-12055; Life Technologies). Samples were taken at the following steps of the cell isolation procedure:
(1) After completion of the digestion process and addition of albumin (2) After the first resuspension of the SVF cell pellet (3) After RBC lysing and filter through 100-μM nylon mesh
A standard curve was generated using CLS-1 at eight different concentration points.
Flow cytometry
Fresh SVF isolates were suspended in a lysing buffer to decrease the number of RBCs (as previously described) and then sequentially passed through a 100- and 60-μm pore filter to reduce the presence of debris and large cell clumps. SVF cells were then resuspended in flow buffer (DPBB +0.2% BSA +0.09% sodium azide; Pharmingen Stain Buffer Cat No. 554657) at a concentration of 1 × 106 cells/mL.
Cells were stained with saturating concentrations of FITC, PE, PE-Cy5, PE-Cy7, AF700, and APC mouse-conjugated anti-human antibodies (glycophorin A [GYPA], CD45, CD31, CD34, CD146, CD66b, CD15, CD64, CD86, and CD206). The cells were dark incubated for 30 min at 4°C. After incubation, cells were washed once with flow buffer and resuspended in 0.25 mL of cold protein-free phosphate-buffered saline (PBS) (for compensation sample) and flow buffer with SYTOX Blue (for sample). Sample acquisition was done on a BD LSR II flow cytometer (3-laser, 13-color configuration) with FACSDiva 6.1.2 software (BD Biosciences). The 488-nm octagon array is capable of collecting up to seven colors plus side scatter (SSC). The 633- and 405-nm trigon arrays can each detect up to three fluorescent colors. The flow cytometer was used at least 30 min after fluidics start-up to allow warm-up and subsequent reliable stabilization of the system. Seven colors were acquired on this three-laser BD LSR II cytometer. Flow cytometer instrument settings were set using unstained cells. Cells were initially gated using forward versus side scatter to eliminate debris. Using SYTOX Blue to exclude dead cells and GYPA to exclude erythrocytes, the remaining SVF cells to be analyzed were considered to be viable, nucleated cells (i.e., non-RBCs). A positive region was established to define positive fluorescence using a same conjugated isotype-matched control antibody. The number of cells staining positive for a given marker was determined by the percentage of cells present within a gate set to <2% positive for the same conjugated isotype-matched control. A minimum of 500,000 events were counted for each analysis. For compensation purposes, a single fluorochrome signal was acquired with cell suspensions. Compensation values were calculated automatically.
Endotoxin/sterility
Bacterial culturing of SVF samples was undertaken without delay. Within a laminar flow hood, a 0.2 mL aliquot of final cell isolates were aseptically inoculated onto tryptone soya agar (TSA) plates using micropipettes with sterile filter tips. The sample was extended over the plate using sterile Digralsky spreaders, and the TSA plates aerobically incubated at 36°C for 72 h. The plates were subsequently examined for colonial morphology. If microbial colonies were present, a Gram stain assessment was completed.
The presence of endotoxin within the final cell isolate resuspension was measured using the Endosafe-Portable Test System (PTS) (Charles River). Samples were diluted in LAL Reagent Water with a ratio of 1:100, and cartridge sensitivity was 1–0.01 EU/mL. Twenty-five microliters of sample was pipetted into all four sample reservoirs of the cartridges and the assays run with Endosafe®-PTS™ according to the manufacturer's instructions.
A subset of SVF isolates was subjected to Gram staining to detect the presence of bacteria. Standard Gram staining methods were used. Samples were centrifuged at 3000 rpm (Sorvall ST 40R Centrifuge; Thermo Scientific) for 15 min to precipitate bacteria. The majority of the liquid phase was discarded and ∼150 μL of remaining supernatant was used to resuspend the pellet by vortex shaker. A 10 μL sample was smeared on a clean slide and heat fixed. All slide rinsing was completed with tap water. The smear was primarily stained with crystal violet for 1 min, rinsed, flooded with mordant (Gram's iodine) solution for 1 min, and again rinsed. The smear was then decolorized with ethanol for 5 s and rinsed. The smear was counterstained with safranin for 1 min, rinsed, and dried on absorbent paper. Several fields on the slide were observed for bacterial organisms under 100 × objective (Zeiss AXIO Imager 2 microscope).
CFU assay
SVF nucleated cells were seeded in triplicate onto a 10-cm Petri cell culture dish at 100 cells/cm2, in DMEM-F12 + 20% FBS. After 24 h, all culture media and nonattached (i.e., floating) cells were removed, and the dish placed in a 5% CO2 incubator at 37°C and 95% humidity for 2 weeks. During this period, half of the medium was removed and replaced at 3-day intervals. On day 14, the entire medium was removed, and the dish washed with PBS and fixed with 4% paraformaldehyde (PFA):PBS 1× for 30 min while stirring gently at 5-min intervals. The PFA was removed, and the colonies twice washed with PBS and stained with 0.5% crystal violet for 5–10 min depending on the desired contrast. Excess dye was removed by washing with distilled water, and the Petri dish allowed to dry at room temperature. The colonies (≥30 cells) were subsequently counted under the brightfield microscope (Zeiss AXIO Imager 2 microscope).
Results
Demographics
SVF-1
A comparative summary of donor information (age, sex, body mass index [BMI], final dry adipose weight) is listed in Table 1. Tissue was harvested from a total of 23 patients (2 male, 21 female) with an average age of 44, ranging 21–70 years. BMI average was 28, ranging 22–37. Processing volumes (weights), as measured by weight of dry adipose tissue remaining after thorough washing of raw lipoaspirate, averaged 252 g, ranging from 97 to 667 g. The average raw yield of viable nucleated cells was 9.55 × 105 cells per gram of dry adipose, with an average viability of 81.5%, ranging 69–87%. The SVF was washed with ammonium–chloride–potassium (ACK) lysis buffer and passed through a 100-μm mesh filter for subsequent flow cytometric testing. After this secondary processing step, the average initial viable nucleated cell yield was 4.79 × 105 cells per gram of dry adipose, with an average viability of 75.1%, ranging 64.5–88.6%.
Our populations were predominantly females with an average BMI of 28. Approximately 1 × 106 viable cells per gram of dry adipose were obtained after dissociation and digestion, regardless of the device.
BMI, body mass index; SVF, stromal vascular fraction.
SVF-2
Tissue was harvested from four female individuals (n = 4) with an average age of 52, ranging from 32 to 68. Initially, one sample was processed in triplicate using three separate SVF-2 devices to validate device protocol and performance. The remaining three samples underwent a single isolation and process procedure. The average BMI was 26.6, ranging from 22 to 34. The average raw yield of viable nucleated cells was 1.006 × 106 cells per gram of dry adipose processed, with an average viability of 82.2%, ranging from 80.6% to 84.0% (Table 1).
Flow cytometry
The resultant data for lipoaspirate samples isolated using the SVF-1 and the SVF-2 devices are reported, allowing direct comparison between the two devices.
As summarized in Table 2, flow cytometric characterization of SVF samples revealed a range of constituent cell subpopulations. Representative flow cytometric plots are presented in Supplementary Figure S1 (Supplementary Data are available online at
Constituent cell subpopulations and corresponding antibody combinations are indicated. Representative flow cytometric plots are available in Supplementary Figure S1. The gray shadings represent root markers used in cell phenotyping.
Although no consensus CD expression pattern for pericytes exists, CD146 is a component of endothelial junctions critical in cell–cell adhesion, and therefore, CD146-positive cells are presumed to be pericytes.
STDEV, standard deviation.
SYTOX Blue dye was used, in combination with forward scatter (FSC) and/or side scatter (SSC), to exclude dead cells and small debris, and glycophorin A (GYPA) to exclude erythrocytes. The resulting live, non-red blood SVF cells were the focus of further analysis. SVF cell components were separated into two fractions according to CD45 membrane expression. In SVF-1 isolates, leukocytes (WBCs), as defined by transmembrane protein tyrosine phosphatase CD45+ staining, constituted ∼14% of total viable nucleated cells (14.13% ± 9.5%, n = 23).
Granulocytes represent ∼1% of the total live cells in SVF-1 cell fractions as evaluated by the expression of CD66b (0.99% of total live cells, n = 23) or CD15 (0.87% of total live cells, n = 21). Nearly 8% of total live cells express the macrophage/monocyte marker CD64 (n = 17). Only 0.91% of total live cells (n = 8) express macrophage type I marker CD86, whereas 5.6% (n = 8) express macrophage type II marker CD206. Similarly, SVF-2 isolates demonstrated a similar trend of low CD86 expression with comparatively higher CD206 positivity, at 0.6% and 4.8%, respectively (Table 3).
Average gated live cell flow results for the SVF-2 device. The gray shadings represent root markers used in cell phenotyping.
The majority of nucleated cells present in the SVF isolates are not leukocytes (i.e., CD45−), regardless of which device is used. Of these, a percentage of cells stain with markers that are suggestive of endothelial lineages. This includes “mature” endothelial cells (CD31+, 9.8% ± 6.4% of total viable SVF-1 isolated cells, n = 23); however, nearly all of these cells also stain for CD34+, supporting their characterization as endothelial progenitor cells (CD31+CD34+, 9.4% ± 6.3%, n = 23). Similarly, SVF-2 isolated viable cells were composed of both mature endothelial cells (CD31+, 7.33% ± 1.63%), the vast majority of which were CD34+ progenitor cells (7.28% ± 1.62% of viable nucleated cells).
The majority of viable cells within the SVF-1 isolates (71.5% ± 11.8% of total viable nucleated cells, n = 23) are neither white blood cells nor endothelial cells (i.e., CD45− and C31−). Within this fraction, about 16% of cells stain positive for CD34, marking the putative adipose progenitor cell (16.3% ± 14.7%; n = 23). The remainder of these CD45−/CD31− cells, however, stain negative for CD34 and make up over half of the viable nucleated cells in SVF (55.6% ± 20.4%; n = 23). When interrogated further, the vast majority of these cells stain positive for the marker CD146 (CD45−/CD31−/CD34−/CD146+: 31.3% ± 21% of viable nucleated cells; n = 23), which is often used to characterize pericyte cells.
In SVF-2 isolates, the majority of cells present were again nonleukocytes, as 79% (79.3% ± 6.2%) demonstrated CD45 negativity (Table 3). Of these cells, the vast majority are also negative for CD31 (CD45−/CD31−; i.e., non-endothelial, nonleukocyte). Within this fraction, ∼18% (13.08% ± 4.84% of total viable nucleated cells) are adipose progenitor cells as determined by CD34+.
CFU assay
After plating at an initial density of 100 cells/cm2, the average colony-forming unit (CFU) (SVF-1 device) was equal to 303 ± 35 versus the average CFU (SVF-2 device) was 274 ± 84 at 2 weeks. These two values are not statistically different (p > 0.05 | n = 3).
Purity
All isolates obtained using the described protocols and devices were negative when analyzed for endotoxin (i.e., <0.01 EU/mL) except one SVF-1 donor sample, whose sentinel cell cultures demonstrated bacterial contamination at day 7. A Gram stain also confirmed the presence of bacteria. Gram stain analysis of the remaining subset of SVF samples isolated from both devices revealed no evidence of bacteria.
Levels of residual collagenase activity were assessed throughout the cell isolation procedure and in the final cell suspension. The starting concentration of collagenase was 14,000 collagenase digestion units (CDU/mL). Standard curve generation yielded a coefficient of correlation of 0.997. At the conclusion of the 40-min incubation/digestion phase and the addition of albumin, the level had decreased 40-fold to less than 350 CDU/mL. After lysing of RBCs and filtration through a 100-μM mesh, the average collagenase level had decreased to less than 10 CDU/mL (Fig. 3A, B).
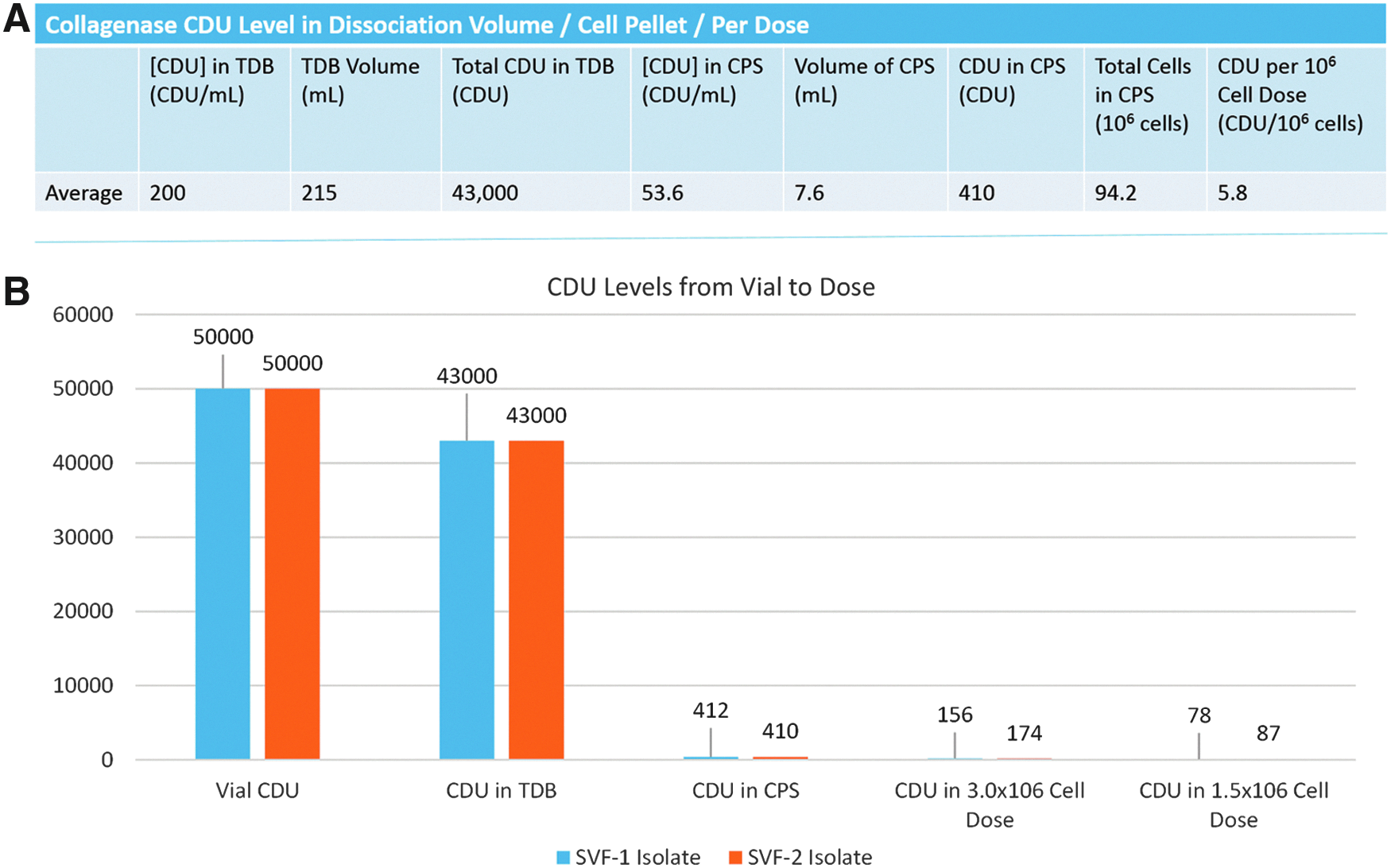
Residual collagenase levels in SVF isolates. SVF sample purity obtained from isolation protocols is critical, particularly in point-of-care therapeutic applications. Both GID SVF devices yield isolates with less than 10 CDU/mL, 1400 times lower than the initial concentration.
Discussion
Over the last decade, adipose tissue has emerged as a premiere candidate source of cells for evolving tissue engineering and regenerative therapies. Adipose tissue was once believed to function as merely padding for internal organs, nerves, and vessels but is now recognized as a complex endocrine organ that impacts a wide array of physiological and pathological processes. In the 1960s, Rodbell described the enzymatic dissociation and separation of cells within adipose tissue, ushering in a new era of study and understanding of the biology and biochemistry of adipose tissue.5,6
In 2001, Zuk et al. published the first peer-reviewed article describing the presence of cells within lipoaspirate with multilineage developmental potential. 7 Studies attempting to verify ASC multipotency were met with skepticism, citing both the extreme in vitro chemical environments required to initiate differentiation and artifactual assays used to verify organ-specific identities.
Despite this, the number of publications related to this field has exponentially increased, with over 1200 publications in 2014 alone. In an effort to address skepticism, many scientists have utilized in vivo modeling. For example, Levi et al. demonstrated that hASCs persisted, proliferated, and osteogenically differentiated in immunodeficient mice with critically sized calvarial defects and intact dura mater. 8 In vivo differentiation has been confirmed in other tissue types, including chondrocytes and hepatocytes.9,10 In addition, many studies indicate that ASCs induce the differentiation and stabilization of host mesenchymal progenitors into target cell types, such as the formation of a vascular network via cotransplantation of ASCs and endothelial progenitors into nude mice. 11 Taken together, ASCs have unique potential for therapeutic applications, regardless of whether these cells are supplying putative stem cells or modifying the chemical micromilieu to induce the proliferation and differentiation of host tissue multipotent progenitors and stem cells.
The growing global interest in the use of adipose tissue as a source of cells, extracellular matrix, and biochemical factors for in vitro and in vivo regenerative and tissue engineering purposes is recapitulated clinically. A search of the NIH clinical trial site,
Currently, there are a number of devices/systems being developed around the world for the isolation of the stromal vascular cellular fraction 12 (Supplementary Table S1). None of these devices is currently approved for clinical use in the United States, although several are being studied in the context of formal IRB- and/or FDA-sanctioned clinical trials. To our review of the literature, few publications exist involving a direct comparison between two or more of these devices. 12
Unlike other devices in development, the GID SVF device platform is a disposable, single-use, closed-system device that is compatible with standard, commercially available bench-top centrifuges and warmer-shakers. For tissue harvest, it can be placed in the sterile field of operation and connected in series between the suction cannula (“upstream”) and a “downstream” waste container. A mesh filter bag within the outer plastic shell retains adipose tissue fragments as liposuction effluent passes through the device thereby ensuring that tissue is never removed from the device during the entirety of the washing, dissociation, and concentration steps of the SVF isolation procedure (Figs. 1 and 2). At the completion of the isolation, the SVF cell pellet is removed directly from the device using a sterile syringe, ready for use or further manipulation (Fig. 2). With experience, the entire process from the time of final tissue harvest to delivery of the SVF cell pellet takes ∼70 min.
In addition, several clinical studies have used the stromal cell isolates obtained from the SVF devices described herein with good therapeutic results. Dos Anjos et al. reported that fat grafts enriched with SVF cells (isolated from the SVF-1 device) significantly decreased early postsurgical breast edema and improved long-term volume retention. 13 Both Garza et al. and Fodor et al. reported that SVF cellular isolates (again isolated from the SVF-1 device) injected intra-articularly significantly decreased osteoarthritic knee pain.14,15 Collectively, these studies speak to the potential anti-inflammatory and reparative abilities of the SVF, despite consisting of <0.1% of putative stem cells. For importance of clarity, this article intends only to describe the isolation, characterization, and (potential) clinical use of SVF cells (i.e., freshly isolated, uncultured cells), as opposed to ASCs (i.e., plated, culture-expanded cells). Indeed, from scientific, therapeutic, and regulatory perspectives, it is important to distinguish the subtle but critical differences between these two distinct cell populations.
The SVF excludes mature adipocytes and encompasses nonlipid-bearing cells, and is therefore easily separated and concentrated by postdissociation centrifugation. Notably, the SVF is a heterogeneous cell population, and the delineation and characterization of specific subpopulations within it remain an area of active interest.1,16,17 As might be expected with the use and analysis of primary human tissue, there is evidence of variability among and within existing reports. There are a number of potential sources for this variability, including but not limited to harvest methodology (e.g., procedural duration, tissue depot or layer, use of tumescence, suction cannula size), cell isolation methodology (e.g., washing solution, dissociative enzyme composition and duration, culture medium), and donor attributes (e.g., sex, age, BMI).17–19 For example, Dos-Anjos Vilaboa et al. have shown that age is inversely correlated with SVF cell yield. 20
There are also many potential sources of variability related to analytical methods, including methods and duration of staining, use of specific hardware/software products (e.g., cell counter, flow cytometer), choice of reagents (e.g., antibodies), sample and reagent temperatures, and all the user biases/errors that are known to occur with such activities. 21 While the use of defined and documented protocols (i.e., standard operating protocols or SOPs) helps to limit many of these analytical sources of variability, they do not necessarily negate those related to inherent biological diversity.
The donors in our series were predominantly female, with an average age of 44 and average BMI of 28. Our methods and results, including the average volume of tissue processed, the nucleated cell yield per gram (or mL) of tissue, and the percent of viable cells, compare quite favorably to similar data reported in the literature, as summarized in Figure 4.22–24

Device performance: comparison of cell yield and viability. Based on information extrapolated from published literature, the GID cell isolation platform (SVF-1/SVF-2) compares favorably to other cell isolation devices. Specifically, it produces large total cell yields on the order of 950,000 cells per gram of adipose processed, with an average viability of 82%. Color images available online at
Regarding cell characterization, the flow cytometric identification of specific cell subpopulations involves the use of putative lineage-specific cell surface markers. The specificity of each lineage-defining protein is related to context, the use of additional markers, and to some extent a “majority consensus” within the peer-reviewed literature. In rare instances, a single cell surface marker can reliably identify a given lineage, such as CD45+ for WBCs and CD206+ for RBCs. In most cases, however, additional markers are required to provide additional support for a given lineage, essentially compiling additive levels of evidence in support of a given phenotypic lineage.
The flow results presented in this study are largely consistent with previous reports of human SVF (Supplementary Table S2).2,16,22–30 We identified a small percentage of RBCs in SVF isolates, which can vary between groups depending on whether washing, centrifugation, hypotonic lysing buffers, density gradients, or a combination thereof is used for red cell removal. The process of RBC lysis and 100 and 60 μm filtration required for flow cytometric analysis significantly reduces the cell yield by ∼50%, as well as the percent viability. Although these secondary processing steps are necessary for SVF flow cytometric characterization, filtration may also be necessary for certain clinical therapies that require intravenous delivery, making the postfiltration yield and viability values of interest.
In our isolates, up to one-third of SVF cells are leukocytes with a substantial portion of these being monocytes/macrophages. A phenotypic and functional dichotomy of the mononuclear/phagocyte cell population has been described in the literature and is determined mainly by the immunologic microenvironment.31–35 In short, an M1 “fight” phenotype is characterized by the inhibition of cell proliferation and the initiation of tissue damage, whereas the M2 “fix” phenotype is associated with the promotion of cell proliferation and tissue repair. 36 Interestingly, our results demonstrate that the vast majority of monocytes/macrophages within SVF populations isolated with the described methods are of the M2 or reparative subtype. In the context of putative stem cells within the SVF, it is also intriguing to note the limited but emerging evidence that suggests that monocyte/macrophage plasticity may extend well beyond the borders of hematological lineages.37–39
The nonleukocyte (CD45−) fraction of SVF can be broadly divided into an endothelial (CD31+) cell fraction, and a nonendothelial fraction (CD31−). Mature endothelial cells are traditionally defined by CD31 staining (in the context of CD45−), and endothelial progenitor cells are further defined by CD34+ staining.40,41 Our results show that the vast majority of endothelial cells isolated using the GID SVF platform and protocol are progenitor cells, accounting for up to 20% of the total live nucleated cells harvested.
The largest cell fraction within SVF isolates is one that stains for neither leukocyte (CD45−) nor endothelial (CD31−) markers. This fraction, which contains the putative mesenchymal stem cell of adipose tissue, can be further characterized using additional cell surface markers, such as CD34. Due to its historical association with (and continued use for) the enrichment of hematopoietic stem cells, CD34 is often thought of and used as a marker of “stemness” for mesenchymal cells also. Although little is known about the specific function of this transmembrane phosphoglycoprotein, there is emerging evidence that demonstrates its repeated association with a wide range of progenitor cells, including those of endothelial, mesenchymal, and epithelial lineage. 42
Similarly, CD146 is another marker that is increasingly used to characterize the adipose-derived SVF.16,43–45 It is a cell adhesion molecule also known as melanoma cell adhesion molecule or MUC18, and was historically used as a marker of endothelial cell lineage. 46 More recently, however, its expression has been associated with the identification of pericytes. The currently held concept that mesenchymal stem cells are pericytes (and/or vice versa) is supported by several independent reports in the literature.47–50 Similar work demonstrates that mesenchymal stem cells that express higher levels of CD146 on the cell surface display greater differentiation potential. 51
While further “dissection” of the SVF population using additional cell surface markers may be worthwhile and useful, the diversity of nature does not fit neatly into the descriptive categories that we have defined at a given time. For example, when four or five cell surface markers are evaluated at once, several subpopulations within the SVF emerge that remain speculative at best—they do not fit neatly into a definitive categorical lineage at this time. Some of these cells may reflect a gradation of developmental states within or between lineages; or they may represent an “activated” versus “nonactivated” state of a given phenotype.
A CFU assay was performed on isolates from both devices. As shown in Figures 5 and 6, the CFU assay results ranged from 2.7% to 6.1%, with an average of 5.34% ± 0.87% and 4.80% ± 1.49% for the SVF-1 and SVF-2 devices, respectively (cell colonies with more than 30 cells per number of seeded cells), consistent with previously published results.19,28,52,53 The CFU analysis is a critical assessment of cell viability, as some SVF production methods may yield viable cells at PoC that produce a decreased number of colonies, 12 or possibly have abbreviated culture periods and decreased proliferation capacities—differences that have been demonstrated in other stem cell sources. 54 Finally, our results show that the isolation of SVF cells using the GID SVF-1 and GID SVF-2 devices and their associated standardized methods is safe, yielding cells that are free of bacterial contaminants as measured by endotoxin levels, Gram stain, and culture. In addition, the levels of residual collagenase at the completion of the cell isolation procedure averaged 10 CDU/mL or less, a value that is 1400-fold lower than the starting concentration. For reference, the therapeutic clinical effects of collagenase for the treatment of Dupuytren's disease require a dose of about 10,000 CDUs in a volume of 0.25 mL, for a concentration of 40,000 CDU/mL.55,56
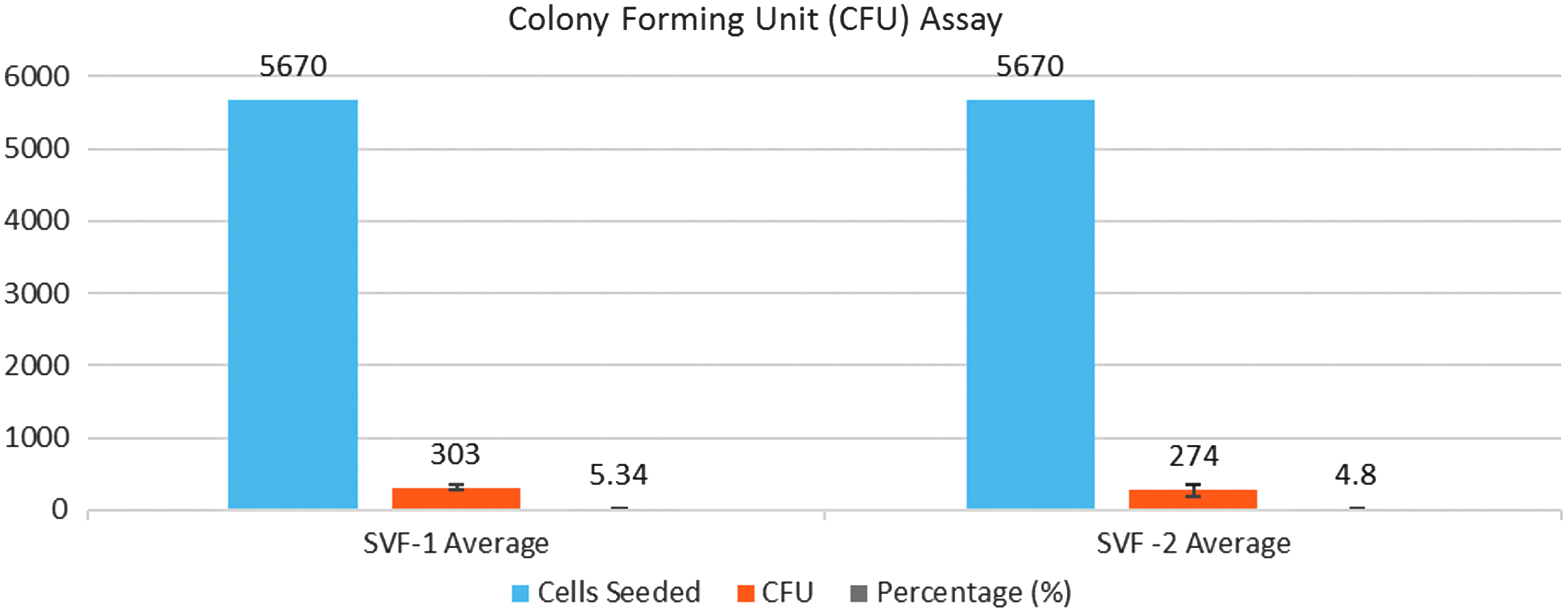
CFU assay results demonstrate an average of 5.34% and 4.80% for the SVF-1- and SVF-2-isolated SVF cells, respectively, and retain the ability to form colonies, consistent with previously reported results.16,25,49,50 Device-specific averages were not statistically different (P > 0.05). Color images available online at
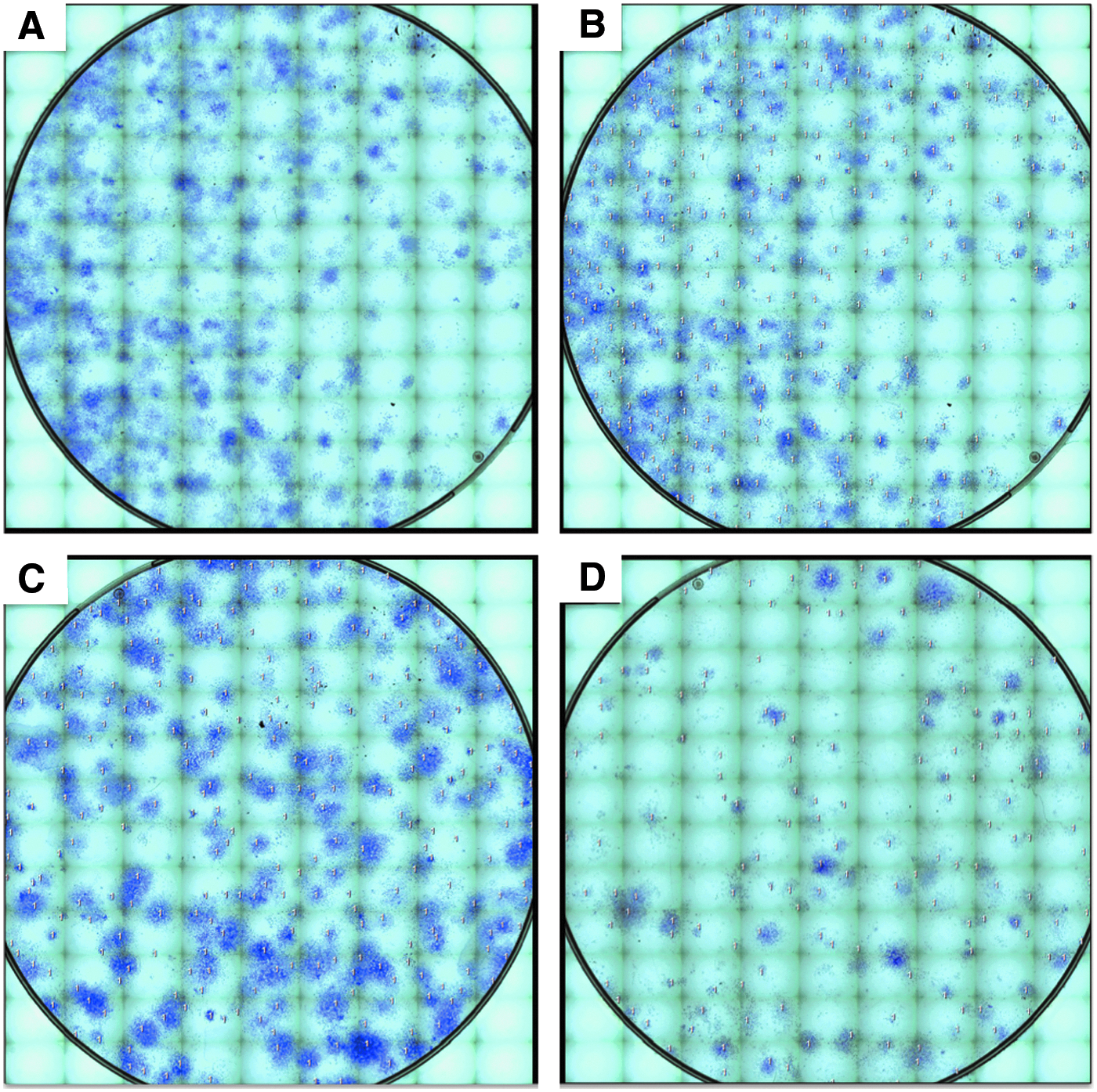
Representative CFU assays from chronological patients. Colonies were defined as a focus of ≥30 cells.
Conclusions
In conclusion, the GID SVF cellular isolation platform produces a reproducible and sterile cell output free of endotoxin and microbial contamination, with defined subpopulations of cells. These cells consist of vascular endothelial cells and associated progenitors, leukocytes, ASCs, pericytes, and putative-lineage committed precursors. In addition, these results are consistent between devices, demonstrating the flexibility of this closed-system platform depending on donor sample size. Finally, the modular nature enables scalable cell “production” for a wide range of tissue volumes, ranging from 125 g of tissue in a single SVF-2 run, to 1200 g in four SVF-1 devices run in parallel (as limited by the centrifuge rotor), thus enabling the clinician to rapidly produce autologous cells for a wide range of clinical indications at the PoC.
Footnotes
Acknowledgments
We thank Ricardo Ungaro for his guidance and assistance with the flow cytometer and associated techniques. In addition, we thank the surgical residents, surgical staff, and nursing staff at the University of Florida Department of Plastic and Reconstructive Surgery for facilitating the harvest, donation, and transfer of patient tissue for this research. This work was supported by the AFIRM I award W81XWH-08-2-0034 to Rutgers University and a subaward to the University of Florida.
Authors' Contributions
J.C.B.: Collection and assembly of data, data analysis and interpretation, manuscript editing and writing, figure and table generation. H.S.: Conception and design, collection and assembly of data, data analysis and interpretation, manuscript writing, figure and table generation. Y.L.: Collection of data, data analysis and interpretation. N.Y.: Conception and design, collection and assembly of data. N.P.: Collection and assembly of data, figure and table generation. A.J.K.: Conception and design, provision of study material, data interpretation, manuscript editing, figure and table editing, and final approval of manuscript.
Disclosure Statement
A.J.K. is a founder, Board Member and owns equity in The GID Group, Inc., a private company that owns the rights to the SVF isolation devices described in this research. No other authors have a conflict to disclose. The research described was partially funded by The GID Group under the auspices of an NSF I/UCRC grant program. The company did not have any oversight over study design, results, or publication.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.