
Abstract
Three-dimensional (3D) imaging techniques provide spatial insight into environmental and cellular interactions and are implemented in various fields, including tissue engineering, but have been restricted by limited quantification tools that misrepresent or underutilize the cellular phenomena captured. This study develops image postprocessing algorithms pairing complex Euclidean metrics with Monte Carlo simulations to quantitatively assess cell and microenvironment spatial distributions while utilizing, for the first time, the entire 3D image captured. Although current methods only analyze a central fraction of presented confocal microscopy images, the proposed algorithms can utilize 210% more cells to calculate 3D spatial distributions that can span a 23-fold longer distance. These algorithms seek to leverage the high sample cost of 3D tissue imaging techniques by extracting maximal quantitative data throughout the captured image.
Introduction
N
Quantitative image analysis has been applied in various systems to determine how spatial tissue organization influences cell activity and fate. Studies include in vivo animal models, centered around intravital imaging at continuous but short (<24 h) durations limited to the few animals or tissues (zebrafish and murine calvarial marrow) able to be imaged while alive,17–21 or in vitro two-dimensional (2D) cell cultures appropriate for studies of spatial organization and cell–cell association.22–24 Most in vivo and in vitro three-dimensional (3D) cultures are not able to implement intravital imaging throughout the growth area of interest and require invasive techniques or culture termination to characterize the formation of cell–environment tissue networks in situ.15–16,25–29 Maximizing extraction of data from these rare, expensive 3D analyses has been a major driver to develop new imaging instruments and techniques rather than the postimaging algorithms that quantitate their output.6–10
Herein, quantitative tools are developed to analyze 3D spatial heterogeneity throughout the entire captured image to maximize data output for expensive, destructive imaging analyses. They work by dividing the full image space into regions, counting the number of cells within each region, and calculating the volume of each region so that regional cell density can be determined. The novel algorithms enable the assessment of complex volumes in regions toward the periphery of the image, thus increasing analysis distance (range) as well as information content (number of cells surveyed). Nonetheless, the proposed 3D analysis represents an estimation whose accuracy is dependent on user-defined inputs. Therefore, results must be validated against controls, such as regions of known volume and data of known density.
Two algorithms to quantify full-image spatial heterogeneity are presented and applied to immunofluorescent (IF) images of a 3D culture system consisting of a synthetic scaffold impregnated with hollow fibers. 30 The first algorithm quantifies the distribution of cellular density from a region of interest and is applicable to recent in vivo studies, including the measurement of oxygen and cellular phenotype, stress, and depiction of regional niches of cell activity and migration.14–19 The second algorithm quantifies cellular association by calculating the distribution of cellular density from each cell imaged. Cell association metrics have been useful to assess roles of cell–cell communication, cell attraction and repulsion, as well as cell colocalization.11–13,23,29 Although current cellular distribution algorithms only analyze a central fraction of imaged data, the method presented herein allows for the utilization of all cells and environmental space imaged.11–13 Specifically, the presented algorithms utilize 3.6-fold more 2D area or 2.5 × 104-fold more 3D volume and survey 1116 cells instead of 235 cells using the 2D analysis or 524 cells using the 3D analysis to help leverage the high cost of tissue culture.
Materials and Methods
Three-dimensional cultures
Mononuclear cells (MNCs) from human umbilical cord blood were cultured in a hollow fiber bioreactor as previously described. 30 In brief, a polyurethane scaffold was formed around ceramic hollow fibers and coated with collagen. Cord blood MNCs were inoculated into the scaffold, whereas hollow fibers were perfused with serum-free medium for 28 days, when a representative scanning electron micrograph to demonstrate bioreactor topology was taken (Fig. 1A).
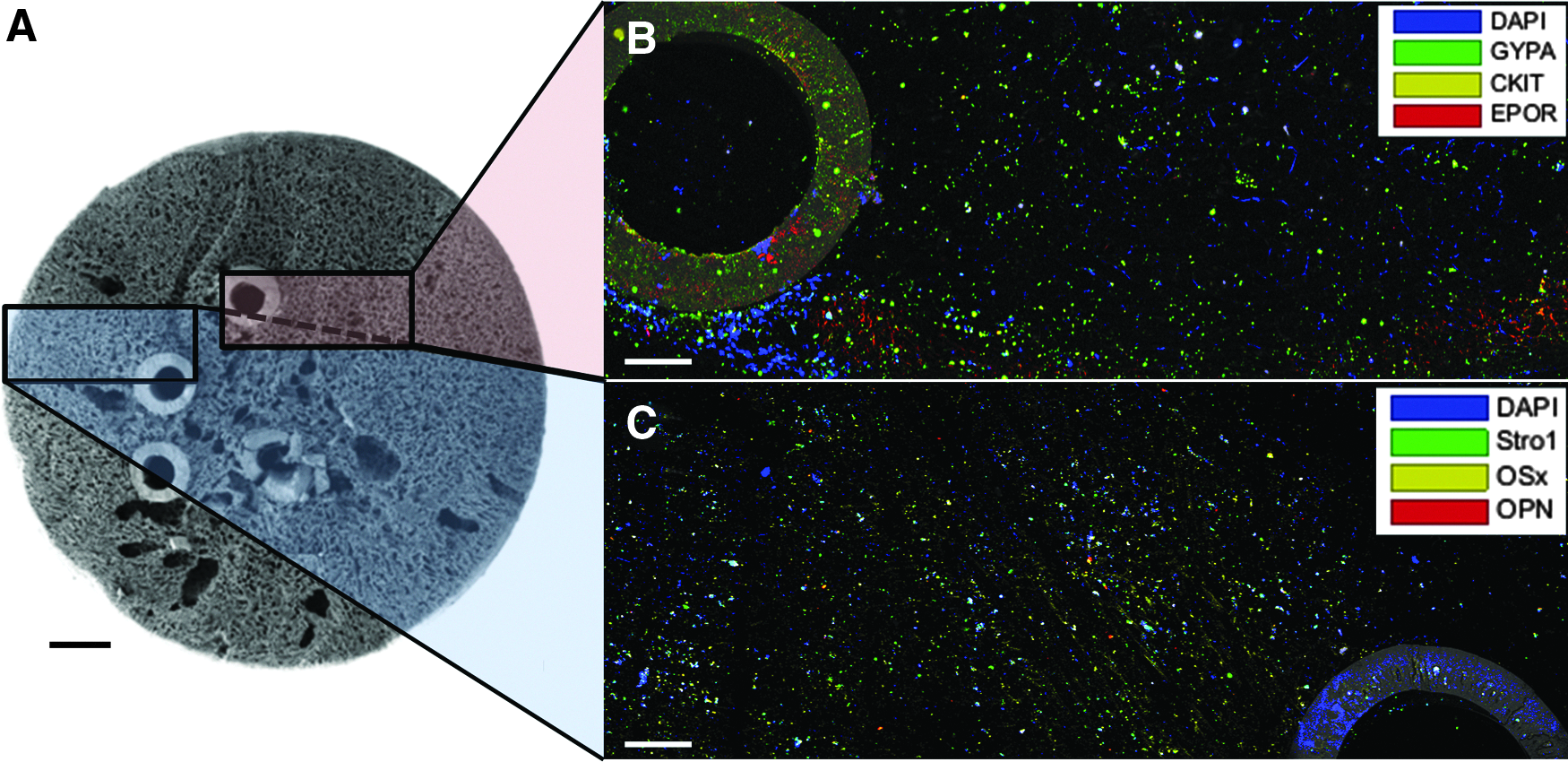
Imaging regions of the 3D culture device.
IF sample preparation
For analyses, the 3D cultures were snap frozen in liquid nitrogen and preserved at −80°C until sectioning. Sectioning occurred at −20°C into thick histological sections of ∼1 mm. The frozen sections were directly fixed in ice cold 4% paraformaldehyde (BDH Laboratory Sciences, Poole, Dorset, United Kingdom) in phosphate-buffered saline (PBS; Life Technologies, Paisley, United Kingdom) overnight followed by a 2-h permeabilization with 0.1% Triton X-100 (Sigma-Aldrich, Poole, Dorset, United Kingdom) in staining buffer (1% bovine serum albumin [Sigma-Aldrich)], 0.5% Tween-20 [Promega, Southampton, Hampshire, United Kingdom], and 0.01% NaN3 [Sigma-Aldrich] in PBS). This was followed by a 4-h blocking step with 10% donkey serum (AbCam, Cambridge, United Kingdom) in staining buffer and overnight incubation with primary antibodies and isotype controls (Table 1; AbCam) in staining buffer at 4°C. Thereafter, 6-h staining was undertaken with secondary antibodies: donkey antirat Alexa Fluor 488, donkey antisheep Alexa Fluor 488 (AF488), donkey antirabbit Alexa Fluor 555 (AF555), and donkey antimouse Alexa Fluor 647 (AF647) all at 1:500 dilution in staining buffer at 4°C (Life Technologies). These steps were followed by a 4′,6-diamidino-2-phenylindole (DAPI) counterstain (Fisher Scientific, Loughborough, Leics, United Kingdom) overnight at 50 μM in PBS at 4°C, and samples were stored in 0.01% NaN3 in PBS at 4°C. Each step was separated by single or multiple 15 min washing steps in appropriate buffer.
All products were obtained from AbCam with product number stated. The two samples presented in Figures 1–5 were prepared alongside appropriate isotype and unstained controls.
IgG, immunoglobulin G.
Confocal microscopy image acquisition
The fixed, stained microscope sections were imaged on a Leica SP5 inverted confocal microscope with Leica LAS AF software (Leica, Milton Keynes, United Kingdom) using 405, 453, 488, 543, and 633 nm lasers and filters for DAPI, reflectance, AF488, AF555, and AF647 stains in two sequences of excitation for two-color collection then three-color collection without any detectable spill over. Images were taken at 512 × 512 pixel resolution using a 10 × dry microscope lens for a resolution of ∼3.03 μm per pixel with Z-stacks acquired in 5 μm slices, selected as the minimum lens magnification to ensure single-cell stain recognition. 19 All laser voltage, filter wavelength, and capture settings were identical for corresponding samples and controls. Each sample was captured as three adjacent 3D images with 7% overlap to allow collation of three individual 1551 × 1551 μm2 square images into a 4437 × 1551 μm2 rectangular image with a depth of 350 μm for Figure 1B and 230 μm for Figure 1C. Unstained and isotype controls were captured under identical conditions. Images were not manipulated.
Cell localization
Once captured, images were analyzed using the Fiji image processing package of ImageJ: (1) the grid/collection package collated together adjacent 3D images, (2) the subtract background package decreased autofluorescence and self-absorbance, and, finally, (3) the 3D object counter package located the center of each individual fluorescence stain, (X,Y,Z) c , which was exported into MATLAB for further analysis (The MathWorks, Inc., Natick, MA).31–33 Representative confocal images are shown in Figure 1B and C in Table 2, with stain localization for Figure 1B shown in Figure 2A and B. The presence of each fluorescent antibody stain, (X,Y,Z) c , was biologically validated in comparison with isotype controls, which contained <1% of detected sample stains. All computational postprocessing was run on a 3.4 GHz machine with 8 GB RAM and an Intel® Core™ i7–4770 CPU. Processing times are provided and correspond as 1 s to 1.2 × 108 random points sampled from a uniform distribution in MATLAB on this machine.
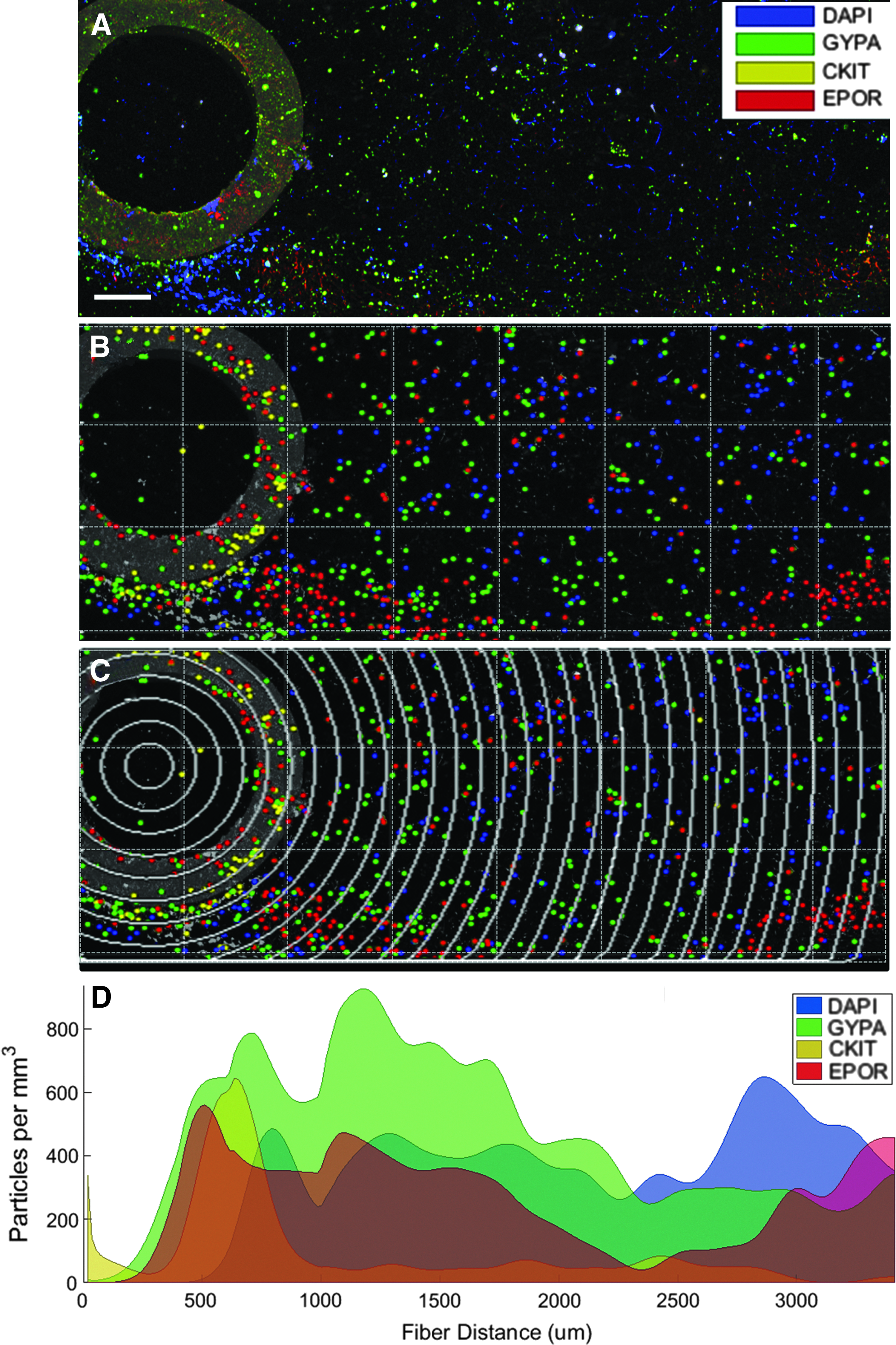
A visualization of the 2D distance cell density analysis.
2D, two-dimensional; 3D, three-dimensional.
Two-dimensional distance distribution
Assessing cell density as a function of distance from an environmental region of interest is useful in examining cellular gradients across tissue sections.14–19
To determine cellular distribution in 2D, 3D confocal image z-stacks (Fig. 1B) were compressed into 2D so that a given line of interest running perpendicular to the image plane becomes a point (X,Y)
a
. Cellular heterogeneity was studied as a function of XY distance from this point of interest (Fig. 2C, D), where individual cell position (X,Y)
c
and its distance from the point of interest (X,Y)
a
were defined as 2D Euclidean distance:
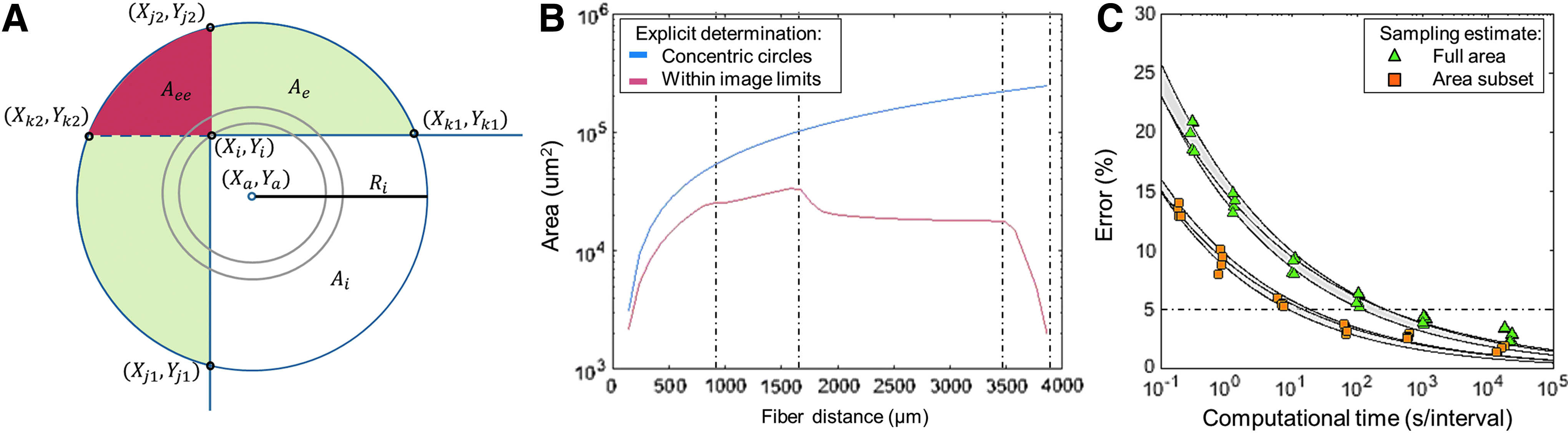
The full image field distance was defined as the distance from the point of interest to the furthest image pixel, Rmax, and was then discretized into bins of radii, which form concentric circles of area
Areas Ae and Aee were arc angles of off-center circle area segments that were analytically calculated through a series of trigonometric identities (refer to Supplementary Data; Supplementary Data are available online at
Two-dimensional tissue distance cell density analysis.
To assess complex 3D geometries that can also be validated against analytical 2D solutions, Monte Carlo methods have been employed to estimate complex geometrical spaces by generating points from a uniform distribution within a measureable region, such as the complete image volume. 34 However, this method may be inaccurate when insufficient points are generated. Therefore, in the 2D case, estimation error was defined as the percentage difference of the Monte Carlo estimation from the analytical solution. As there was no analytical solution for many 3D cases, Monte Carlo convergence was defined as the percentage difference between estimated Monte Carlo solutions when 104 additional uniformly generated points were appended. 35
The Monte Carlo method was validated for the typical 2D distance distribution (Fig. 3C) by calculating the average estimation error and Monte Carlo convergence across four image geometries, each broken into 40 equidistant intervals from a point of interest. An estimation error of 2.75% from the explicit calculation was reached for 10−6% Monte Carlo convergence in iterations of 104 points, but required 8.6 min. A 10−2% Monte Carlo convergence, which corresponded to an average of 2.4 × 106 generated points or 0.02 s per distance interval with estimation error less than 5% in the 2D distance distribution, was utilized for all subsequent distance distribution analyses.
3D distance distribution
The analysis of 3D cell distributions away from a specific point of interest is often utilized for cell colocalization studies, but is currently limited to cells either near the center of the image or only for very close cell proximities, creating regional bias in analysis.11–12,23,29 To analyze 3D cell distribution from a point of interest
Therefore, cell density within neighborhood
where VL is the full image volume and the concentric neighborhood volume of the prior radial interval Ri−1 is given by Vi−1. This process of uniform point generation and volume calculation was repeated and appended until iterations of calculated Vi vary less than a chosen Monte Carlo convergence limit, as already discussed. The estimation of a 100 μm cell neighborhood (100 μm, 4.2 × 10−3 mm3) inside a large image volume, VL (2.4 mm3), required sampling 5.8 × 107 uniformly distributed random points and 0.49 s, or 9.1 min if repeated for the 1116 cells within Figure 1B.
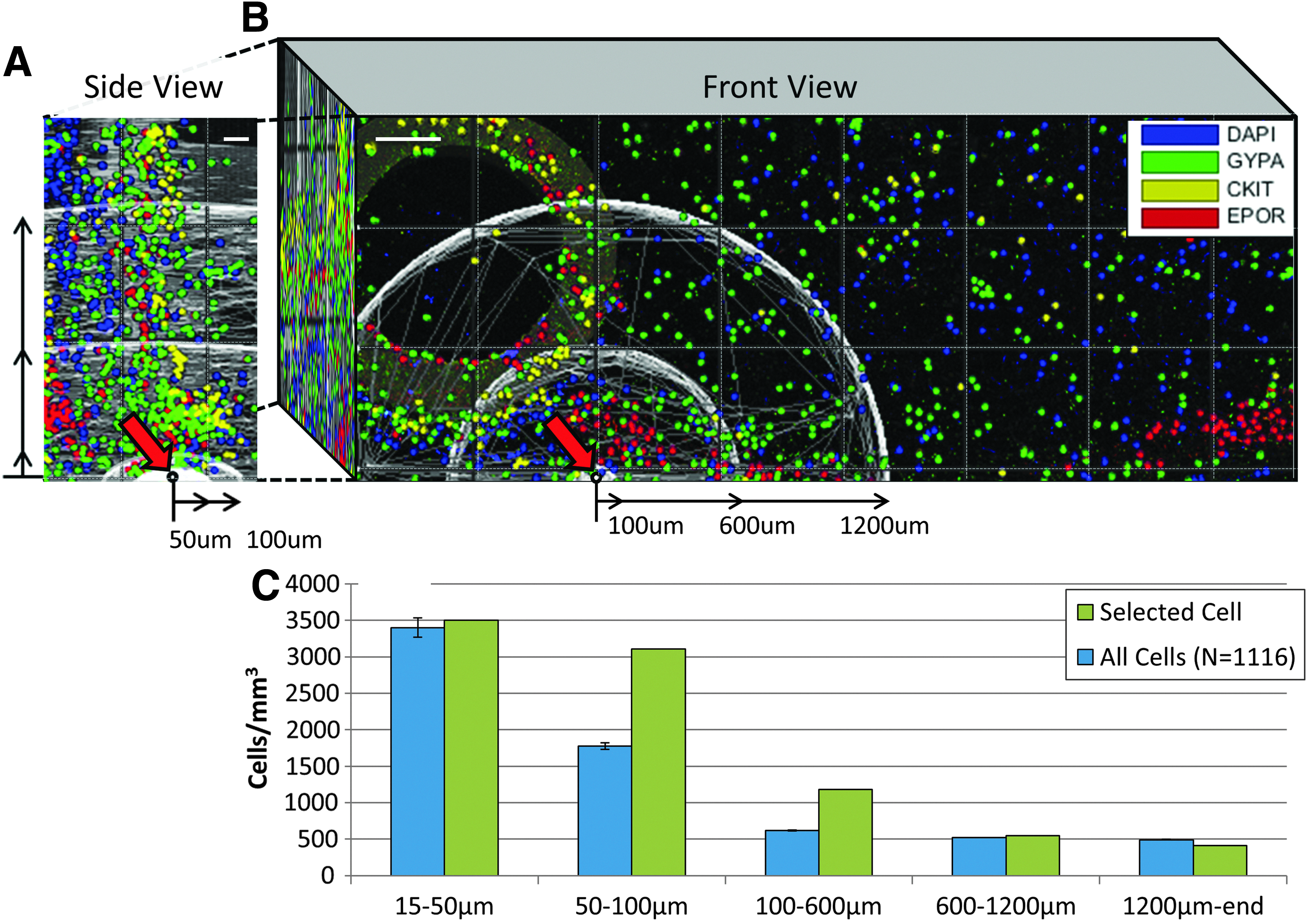
A visualization of the 3D cell clustering density analysis in confocal images.
Computational time decreased by 96% from 9.1 min to 22 s when Monte Carlo point sampling methods were applied within small regions around each cell rather than across the entire image space. A smaller cuboid of known volume, VLS, was constructed to exactly encase the cell neighborhood [volume (2Ri)
3
centered on cell
Fewer sampled points were required within VLS than VL to reach the same Monte Carlo convergence: 2.4 × 106 uniformly distributed random points required 0.02 s per cell neighborhood, or 22 s for all 1116 cells shown in Figure 1B. Using the proposed method, 3D cell density distributions can be quickly analyzed at distances up to half the diagonal image distance, or
Alternatively, an equally spaced 3D grid of k points across the image volume, VL, or a similar image subvolume, VLS, can be used to estimate cell neighborhoods. However, iterative grid generations must be performed with
Statistics
Quantitative results are represented as mean ± standard error of four replicate experimental samples prepared and assessed identically (Fig. 1B), or four replicate computationally generated samples as stated.
Results
Enhanced utilization of imaged data
The presented algorithms completely assess 2D and 3D cell density distributions of imaged histological data, whereas current methods disregard data near image boundaries.11–13 Therefore, the algorithms herein enhance utilization of image data in two ways: (i) by surveying more points of interest and (ii) by probing further distances from each point of interest. With current approaches, an ideally centered point of interest within Figure 1B could only be inspected up to a maximum 2D distance of 775 μm (the closest image boundary); in contrast, using the presented algorithms, a 2D distribution of at minimum 2300 μm (the furthest imaged pixel) can now be calculated from any point within the image.
Cell density distribution algorithms are typically repeated for multiple points, or cells, of interest. A conservative comparison with current methods was performed by analyzing 3D cell density distributions within a 100 μm neighborhood around every cell to demonstrate cell-to-cell association as shown in Figure 1B (similar to Bjornsson et al. 11 ). Current methods utilize only cells further than 100 μm from an image boundary as points of interest (N = 524), whereas with the presented approach, all imaged cells were utilized (N = 1116) at a 23-fold greater distance. The inclusion of these cells improved cell density estimations by 26% and 23% within 15–50 and 50–100 μm intervals, respectively.
Accurate manual scoring allows for the exact measurement of cell positions and cell-to-cell distances, but would be infeasible to manually pinpoint thousands of cells in a large image and infeasible to measure the hundreds of thousands of intracellular distances and the volumetric neighborhoods to calculate cellular density distributions within. The proposed algorithm was compared to manual scoring of random fractions of the entire image shown in Figure 1B, which revealed that partial manual scoring reduced accuracy because of the nonuniform distribution of cells and was unable to analyze cell distributions at long distances as described in the Supplementary Data, while still reducing analysis time from 30 min down to 1 min.
Random sampling accuracy
The point sampling algorithm was validated by assessing uniformly generated random cell distributions, as shown in Figure 5A. These data sets comprised four types of 500 computationally generated cells, which were uniformly distributed within a 4437 × 1551 × 230 μm3 space to produce a known cell density while replicating the number of cells and image geometry of experimental samples, as performed in current studies.14,15 The sampling method accurately analyzed the uniform distribution of cells, where 2000 uniformly generated cell positions were found to have an average of 1257.6 ± 44.8, 1244.8 ± 22.4, and 1297.2 ± 2.0 cells/mm3 in 15–50, 50–100, and 100–600 μm neighborhoods of all other cells, and 1297.0 ± 0.2 cells/mm3 throughout the remaining 2300 μm of the image space (n = 2000 cells). Small 50 and 100 μm neighborhoods contained significantly smaller number of cells, and may have accounted for their higher variability, but even so the density of cells was found to closely approximate a uniform distribution of 2000 cells in the image volume of 1.54 mm3.

Assessment of methods and applications for calculating 3D cell clustering density.
Three-dimensional distribution with regional asymmetries
Many histological images contain gaps or spaces without cells, such as vessels, areas of decalcified bone, or the edge of the sectioned sample. These voids may underestimate cell density during computational analysis if they are averaged into dense cellular neighborhoods. However, if a significantly larger number of cells could be assessed with full image field analysis, regional asymmetries might be more accurately determined. Cell clustering density analyses were run on an identical image, including or excluding regions with engineered void space (Fig. 5B, C), and showed that the region containing the voidage played a relatively minor role in influencing paracrine cell–cell niche communication, but a much greater regional effect correlating with its inclusion or exclusion from the 2D distance cell density analysis. There was a 7.8%, 11.1%, and 44.6% difference in cell density in 15–50, 50–100, and 100–600 μm neighborhoods between the image with and without the fiber, and a 46.2% difference in cell density throughout the rest of the image, demonstrating that this regional phenomenon altered cell localization at large 2D tissue scales, but cell association at close intercellular distances remained consistent throughout the image.
Discussion
Three-dimensional image analyses enable the acquisition of information on spatial cell and tissue heterogeneity because of their quantitative algorithms.6,7 However, current calculations of cell distributions are often constrained to a central subset of the image provided, and are further limited when phenomena of interest are off-center or replicate events per image are assessed. When the entire tissue volume of interest is infeasible to image, recent studies have compensated by quantitating images of incomplete tissue subsections either through normalization against uniformly generated random data within a similar tissue topology13–15 or by only analyzing phenomena that exist within the most central subsection of the image.11–12,16 The former method only provides relative quantitation of images, whereas the later discards nearly half of 3D imaged data.11,12 The presented analyses in this study quantitate 2D and 3D distributions of phenomena throughout any full image, providing a minimum 50-fold increase in analytical range (increased distance times increased cells analyzed) throughout coupling complex Euclidean metrics with Monte Carlo point sampling estimations.
The analysis of cell distributions can be parametrically tuned for computational speed and analytical resolution to avoid miscalculation. Analytical resolution is dependent on the number of regions the image is partitioned into and, for 3D distributions, the Monte Carlo simulation is also dependent on the quantity of uniformly generated random points. The partitioning of image regions is a balance of precision, where larger, coarser partitions may average local spatial phenomena into a larger population of cells, whereas smaller, finer partitions could provide misleading cell densities because of the sharp geometrical discontinuities that exist at image boundaries and the small number of cells that exist within small partitions. The partition balance is observed as the measurement of small cell proximal 3D regions held larger error (4% standard error at 0.0042 mm3) than larger, cell distal 3D regions (0.015% standard error at 2.4 mm3). The generation of sufficient quantities of uniformly generated random points to estimate complex 3D volumes is dependent on system and user-specific error tolerance; in the presented case, an average of 2.4 × 106 points were generated, which yielded a 5% error when compared against known controls, as already discussed.
Many studies of cell recognition and cell–cell or cell–tissue colocalization have avoided the need for efficient image analysis algorithms by capturing multiple or larger images at lower magnification, increasing the cost and time of these already demanding techniques. Morales-Navarrete et al. 36 proposed imaging at a moderate resolution (25 × , 1 μm3 voxel), then retiling parts of this low-resolution image with high resolution (63 × , 0.03 μm3 voxel) to capture both vasculature of ∼100–300 μm diameters for murine liver reconstruction followed by cellular and subcellular reconstruction of hepatocytes. Studies by Khorshed et al. 19 and Sundaramurthy et al. 37 enumerated intravital murine hematopoietic cells at moderate resolutions (20–40 × , 0.2–7.3 μm3 voxel) to inspect intracellular and intercellular activity for small populations of ∼25 cells. The presented study depicts cells at significantly lower resolution (10 × , 46 μm3 voxel), as an optimal balance between image volume analyzed at a tissue scale while still capturing many hematopoietic cells (1000's of 500–33,500 μm3 cells) at high enough resolution to pinpoint cell nuclei and associated fluorescent markers among their environment. This lower magnification and resolution has also been employed in semiautomatic cell and fluorescent marker identification in 3D tissue cultures 38 to assess megakaryocyte distance from vascularization in murine bone marrow, 39 and used as standard images to compare cell motion tracking algorithms across 14 laboratories. 40
The quantitative evaluation of cellular density and heterogeneity is frequently studied and used in vitro and in vivo 3D applications inspecting roles of cellular and environmental interactions. Many 3D culture systems employ surfaces, scaffolds, carriers, or fibers as an aim to impart spatial heterogeneity to mimic and, upon implantation, integrate with many tissue asymmetries present physiologically.1–4,25–28 Pinpointing cell niche placement and cell association as a tool toward developmental understanding, tracking disease progression, and mammalian cell culture bioprocess design has become increasingly studied in ex vivo analogues; however, few intravital analyses have been performed until recently, and none in the adult human.14–29,36–40 Cell colocalization algorithms frequently inspect small distances from cells or tissues of interest such as the effect of high oxygen tension on cells within 150 μm of a vascular sinus and the migration of cells within 100 μm of bone surfaces.16,19 The assessment of spatial heterogeneity away from more complex regions of interest, such as uneven bone surfaces or noncircular vessel walls, could utilize these proposed algorithms to capture the full image field in one of two ways: in an identical method if these phenomena can be described as a function, or through an iterative process once the complex phenomena of interest have been meshed or mapped similar to the detection of the murine marrow bone surface by Khorshed et al. 19
These algorithms are capable of being applied to any quantitative 3D imaging platform, including magnetic resonance imaging (MRI), computed tomography (CT), and optical imaging (UV, visible, IR) because of their use of spatially “tagged” contrast agents or probes (e.g., nanomaterials, labeled small and large molecules, and fluorescent proteins), where discrete cells, phenomena, or regions of interest can be spatially identified.6,7 MRI and CT imaging allow for full specimen penetration but lack multiplexed probe detection, have poorer sensitivity, and lower resolution; only recent advancements have allowed imaging at single-cell resolution. 6 Optical imaging allows for the quantitative analysis of a wide variety of cell–environment interactions and can characterize multiple phenotypes, organelle, and matrix proteins simultaneously by implementing light microscopy with immunohistochemistry or IF imaging of probes and chemicals delineating cell or tissue type, status, and their interactions to quantify the effects of the tissue environment on cell differentiation, proliferation, and stimulation spatially at multiple aspect sizes.8,9 The versatility of optical imaging is frequently implemented to determine quantitative colocalization from the subcellular protein-scale up to multicellular and tissue scales.10–28
The complete quantitative utilization of 3D imaged space is required to leverage the high cost of native or engineered tissue histology. The study of spatial heterogeneity in tissue constructs has become increasingly popular from both advancements in 3D culture systems to mimic physiological structures and functions and advancements in imaging technologies able to better define the performance of natural and engineered cellular environments. The presented image analyses, for the first time, pair Euclidean metrics with Monte Carlo estimations to determine colocalization across the entire imaged data set, assessing 2.2-fold more area per frame in 2D distance metrics to depict cell populations in more distant, peripheral imaged areas from phenomena of interest. In 3D distance metrics, all cell neighborhoods can be analyzed to half the maximum image length, whereas basic algorithms may only assess one perfectly central cell per image; hence, these tools provide 102–104 more replicate cell measurements per image at a similar, if not greater, scope. The fabrication, culture, and analysis of tissue -engineered and clinical samples for image analysis represent a time-, labor-, and cost-intensive process, and efficient post-analysis tools to quantitatively assess the full image field, as presented here, are needed to detail cellular interactions among their environment.
Footnotes
Acknowledgments
This work is supported by ERC-BioBlood (No. 340719), the Richard Thomas Leukaemia Research Fund, and a Royal Academy of Engineering Fellowship to R.M. M.C.A. is grateful for the Imperial College Chemical Engineering Scholarship. The authors are grateful to Deborah Keller and Stephen Rothery of Imperial College's Core FILM Facilities for the useful discussions.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.