
Abstract
Previous studies have demonstrated that extracellular matrix (ECM) can be used in tissue engineering due to its bioactivity. However, adipose-derived ECM (A-dECM) has never been applied in bone tissue engineering, and it is unknown whether it would be beneficial to the growth of bone marrow mesenchymal stem cells (BMSCs). In this study, we produced chitosan/gelatin/A-dECM (C/G/A-dECM) scaffolds via lyophilization and crosslinking; chitosan/gelatin (C/G) scaffolds were used as controls. For the C/G/A-dECM scaffolds, the average pore size was 285.93 ± 85.39 μm; the average porosity was 90.62 ± 3.65%; the average compressive modulus was 0.87 ± 0.05 kPa; and the average water uptake ratio was 13.73 ± 1.16. In vitro, A-dECM scaffolds could promote the attachment and proliferation of BMSCs. In the same osteogenic-inducing reagent, better osteogenic differentiation could be observed for the C/G/A-dECM scaffolds than for the C/G scaffolds. Thus, we conclude that A-dECM is a promising material and that C/G/A-dECM scaffolds are a candidate for bone tissue engineering.
Introduction
E
Extracellular matrix (ECM) contains collagen, elastin, and proteoglycan, which play key roles in cell proliferation, differentiation, and migration and the interactions between cells. 3 In addition, there are several growth factors present in the ECM, such as vascular endothelial growth factor (VEGF) and transforming growth factor-β1. 4 For ECM without cells, both autologous and xenogenous applications are safe. 5 ECM from bone, cartilage, and adipose tissue has been used due to its bioactivity. Bone-derived ECM can be applied for bone repair and regeneration, 6 while cartilage-derived ECM is applied for cartilage regeneration 7 and adipose-derived ECM (A-dECM) is applied for adipose tissue.8–10 However, the availability of bone and cartilage in the body is limited, and damage is inevitable. In contrast, adipose tissue is abundant in the body and easy to obtain.
A previous study confirmed that A-dECM not only promotes the proliferation and differentiation of adipocytes but is also beneficial for the attachment and proliferation of human skin fibroblasts, human aortic smooth muscle cells, chondrocytes, and umbilical endothelial cells. 11 However, the effects of A-dECM on bone marrow mesenchymal stem cells (BMSCs) are unclear, and it therefore remains unknown whether it is suitable for use in bone tissue engineering.
Chitosan is the only cationic polysaccharide with osteogenic properties from natural origin, 12 and gelatin is anionic. The two substances are biocompatible and exhibit low toxicity. Chitosan/gelatin (C/G) scaffolds display satisfactory mechanical properties and have been applied in bone,12–15 cartilage, 16 and soft17,18 tissue engineering. For ECM scaffolds, the compressive strength is low, 4 and associating C/G with ECM can therefore theoretically improve not only the bioactivity of C/G scaffolds but also the strength of ECM scaffolds, while simultaneously maintaining the osteogenic ability of chitosan.
This study focused on a new tissue engineering scaffold containing chitosan, gelatin, and A-dECM and investigated whether it could be a candidate for use in bone tissue engineering.
Materials and Methods
Rats and adipose tissue
S-D male rats (weighing ∼50 g) were supplied by the animal center of the Second Affiliated Hospital of Harbin Medical University. All animal experiments were carried out in compliance with the Institutional Animal Care and Use Committee of Harbin Medical University.
Adipose tissue was supplied through cardiac surgery, obtained from the subcutaneous chest tissue of patients. Patients were informed about and agreed to take part in this study.
Isolation and culture of BMSCs
BMSCs were isolated and cultured as previously reported. 19 Briefly, S-D rats were euthanized via an overdose injection of pentobarbital and their bilateral femurs were then removed. The bone marrow was flushed from the femurs with Dulbecco's modified Eagle's medium (DMEM) (HyClone, USA). After centrifugation, the sediment was resuspended in DMEM +10% fetal bovine serum (FBS) (HyClone) +1% penicillin/streptomycin (Beyotime, Shanghai, China) and then cultured in culture bottles (37°C, 5% CO2 atmosphere, 95% humidity). Two days later, the nonadherent cells were removed, and the adherent cells were further cultured until 80–90% confluence and then passaged. The third passage cells were collected and tested as reported previously. 20 Differentiation of osteogenesis, adipogenesis, and chondrogenesis (Cyagen, Guangzhou, China) was conducted, and immunophenotype was analyzed with flow cytometry.
Preparation of A-dECM
Decellularization was conducted as reported previously. 21 Adipose tissue, cut into pieces, was rinsed with phosphate-buffered saline (PBS) to clean off the blood, then immersed in Tris buffer, frozen at −80°C for 5 h, and rewarmed to 37°C over five freeze–thaw cycles. The pieces were subsequently immersed in 0.25% trypsin/0.01% EDTA (Solarbio, Beijing, China) for 6 h, rinsed three times with PBS, immersed in isopropanol (Xilong, Guangdong, China) for 36 h, rinsed three times with PBS, immersed in 0.25% trypsin/0.01% EDTA for 6 h, rinsed three times with PBS, treated with DNase I and RNase A (Sigma, USA) for 16 h, rinsed three times, immersed in isopropanol for 8 h, and rinsed three times to obtain A-dECM (Fig. 1a). The A-dECM was subsequently lyophilized and stored at −80°C for further use.

A-dECM and A-dECM suspension.
Fabrication of C/G/A-dECM scaffolds
Chitosan (Biosharp, Anhui, China) was dissolved in 1% v/v acetic acid (Tian Li, Tianjin, China); gelatin (Biosharp) was dissolved in double distilled water (ddw) in a 50°C water bath; and an A-dECM suspension was produced using a tissue homogenate machine (Fastprep 24; MP Biomedicals, USA) (Fig. 1b). These three solutions were then mixed to form 100 mL of a homogeneous solution containing 2% w/v chitosan, 2% w/v gelatin, and 1% w/v A-dECM, which was then crosslinked by adding 0.25% v/v glutaraldehyde (Tian Li) overnight. The mixed solution was transferred to a 12-well culture plate, frozen at −80°C for 8 h, and then lyophilized for 48 h to harvest the scaffolds. To remove acetic acid and glutaraldehyde, the scaffolds were immersed in 1% w/v NaOH (Quan rui, Liaoning, China) for 30 min, flushed with ddw, immersed in 2% w/v sodium borohydride (Shan Pu, Shanghai, China) for 30 min, flushed with ddw, and lyophilized to create the final scaffolds (Fig. 2a).

General and internal structure.
Fabrication of C/G scaffolds
Based on the method described above, the mixed solution containing 2.5% chitosan and 2.5% gelatin was lyophilized to harvest the scaffolds (Fig. 2b). Both scaffolds were sterilized with ethylene oxide before further characterization.
Structural performance
We conducted a visual inspection of shape, color, and basic structural characteristics and performed H&E staining to assess the internal structure. Briefly, the scaffolds were immersed in ddw overnight, then dehydrated through an alcohol gradient, and immersed in n-butyl alcohol for 12 h. The scaffolds were subsequently immersed in xylene and liquid paraffin, and then embedded with paraffin. The obtained paraffin block was cut into slices of ∼5 μm and subjected to H&E staining. We conducted a scanning electron microscopy (SEM) analysis of internal structure. The scaffolds were cut and coated with gold palladium. Morphology was visualized via SEM (Hitachi, S-3400n, Japan). Under an appropriate magnification, we measured the sizes of 10 pores randomly selected from 3 random views, and the provided scaffold pore size is the average value. 22
Porosity
Based on a method reported previously, 23 the scaffolds were cut into ∼1 cm3 cubes. Their length, width, and height were then accurately measured with a digital caliper, and the volume (Vw) was calculated. The weight (Ws) was also measured. Next, the scaffold was immersed in a container of ethanol (ρ), and the ethanol displaced by the scaffold was retained in another container. The mixture containing the ethanol and scaffold and both containers were then weighed (W1). After removing the displaced ethanol, the mixture was weighed once again (W2). The porosity was calculated as e = 1−(W1−W2+Ws)/(ρx Vw) × 100%.
Water uptake ratio
The scaffolds were weighted (W1), immersed in ddw for 2 h, and weighed again (W2). The water uptake ratio was calculated as (W2−W1)/W1 × 100%.
Compressive strength
A universal mechanical tester (Instron 5943) with a load cell of 10 N was used. Scaffolds that were 6 mm in diameter and 2 mm in thickness were loaded onto the specimen stage. The scaffolds were compressed at 50 μN/min until they failed to sustain the strength. The compressive stress–strain curve was linear until a strain rate of 50%, and the compressive modulus was at the 50% point.
In vitro cytotoxicity
The scaffold was immersed in 5 mL of medium (DMEM+FBS+penicillin/streptomycin) in a culture bottle (4°C) for 1, 3, or 7 days. We removed the scaffold and collected the mixture containing the medium eluted for cell culture. BMSCs were cultured at a density of 1 × 104 cells/well in a 96-well plate with medium+C/G/A-ECM eluate, medium+C/G eluate, or medium alone. The cells were analyzed at 3 days with a CCK-8 assay kit (Jiancheng, Nanjing, China) according to the manufacturer's instructions. Optical density (OD) values represented absorbency of cells in 450 nm, and the higher the absorbency was the more the cells were. So, cell proliferation ratios were compared by OD values. This procedure was repeated three times for each well.
Cell adhesion
Scaffolds were cut into cylinders with a height of 0.5 cm and immersed in medium for 2 h for pretreatment. The scaffold was mixed with 3 mL of medium with 1 × 105 BMSCs and vibrated overnight on a shock table for dynamic attachment on C/G/A-dECM scaffolds and C/G scaffolds. Cell-seeded scaffolds were removed and further cultured. Then, the nonadherent cells remaining in the medium and the adherent cells were counted (n1). The cell adhesion ratio for scaffolds was (1 × 105−n1)/(1 × 105) × 100%.
Cytoactivity
The viability of cells on scaffolds was analyzed via live/dead staining (Tian Kai, Tianjin, China) and SEM. Cell-seeded scaffolds were further cultured in medium, and the medium was refreshed every 2 days. After 1 and 7 days, live/dead staining was performed according to the manufacturer's instructions. Briefly, a kit contains two reagents, calcein AM and EthD-1. When cells are cultured in mixed liquor, which contains FBS, calcein AM, and EthD-1, calcein AM enters into live cells and combines with special enzyme. Calcein fluorescent molecule stays in live cells, so live cells appeared green. EthD-1 cannot pass the cytomembrane, but it can enter into dead cells and combine with gene segment to form fluorescence, so dead cells appeared red. At 7 days, SEM was performed as mentioned above for the observation of cell attachment on scaffolds.
Alkaline phosphatase
For osteogenic induction (OS) of both scaffolds, cell-seeded scaffolds were cultured with and without OS medium (Cyagen). The medium was refreshed every 2 days. At 7, 14, and 21 days, alkaline phosphatase (ALP) was extracted from the cells as described below. A cell-seeded scaffold was immersed in 2 mL of 0.25% trypsin (Beyotime) for 3 min, and 2 mL of medium was added to neutralize the trypsin. The scaffold was removed, and the mixture, which contained trypsin, medium, and cells, was collected. The mixture was then centrifuged, and the sediment was resuspended with 1 mL of medium. The cell membranes were subsequently disrupted using ultrasound (SONICS Vibra Cell VCX 105, USA). OD and U values were detected and calculated according to the ALP manufacturer's instructions (Jiancheng). This procedure was repeated three times.
Real-time quantitative reverse transcription-polymerase chain reaction
Cells were subjected to extraction as outlined above. Total RNA was isolated using the TRIzol reagent (HaiGene, Harbin, China). Complementary DNA (cDNA) was synthesized with the Golden 1st cDNA Synthesis Kit (HaiGene). Real-time PCR assays for the messenger RNA (mRNA) were performed in a Mini-Opticon2 system (MJ) using Golden HS SYBR Green qPCR Mix (HaiGene) under the following conditions: 95°C for 15 min, followed by 45 cycles of 95°C for 10 s and 60°C for 30 s. To amplify the target genes, primers were purchased from GenScript (Nanjing, China) (Table 1). The gapdh qPCR primer was obtained from HaiGene. Quantitative normalization of the cDNA in each sample was performed using the gapdh gene as an internal control, and the target gene expression levels were calculated using the ΔΔCT method and normalized to the data of the control group. The experiment was repeated three times.
Mineralization deposition
Alizarin red staining (Cyagen) and von Kossa (Leagene, Beijing, China) staining were carried out to detect calcium nodules. After 21 days in medium with or without OS, the scaffold was immersed in 4% neutral paraformaldehyde (Boster, Hubei, China) and flushed with ddw. For alizarin red staining, the scaffold was immersed in alizarin red for 3 min and flushed with ddw to remove redundant dye. For von Kossa staining, the scaffold was immersed in 5% von Kossa reagent, exposed to ultraviolet rays for 10 min, and flushed with ddw. Finally, the calcium nodules were observed with an inverted microscope.
Statistical analysis
In vitro, cytological examination was performed on six replicate scaffolds unless otherwise indicated, and the data are presented as the mean ± standard deviation. Statistical differences were analyzed using independent-samples t-test and one-way analysis of variance, and p-values of <0.05 or 0.01 were considered significant.
Results
Culture and identification of BMSCs
After culturing for 3 days, the medium was replaced and cells attached to the wall of culture bottle. After 7 days, cell confluency reached 80–90% and were passaged. The third-passage cells appeared shuttle shaped and osteogenic, adipogenic, and cartilage induction was as shown (Fig. 3a–d). Immunophenotype was as shown (Fig. 4), and CD29, CD90, and CD44 exceeded 95%, whereas BMSCs did not express CD45, which is marker of hematopoietic stem cells. So, cells met the criteria for mesenchymal stem/stromal cells. 24
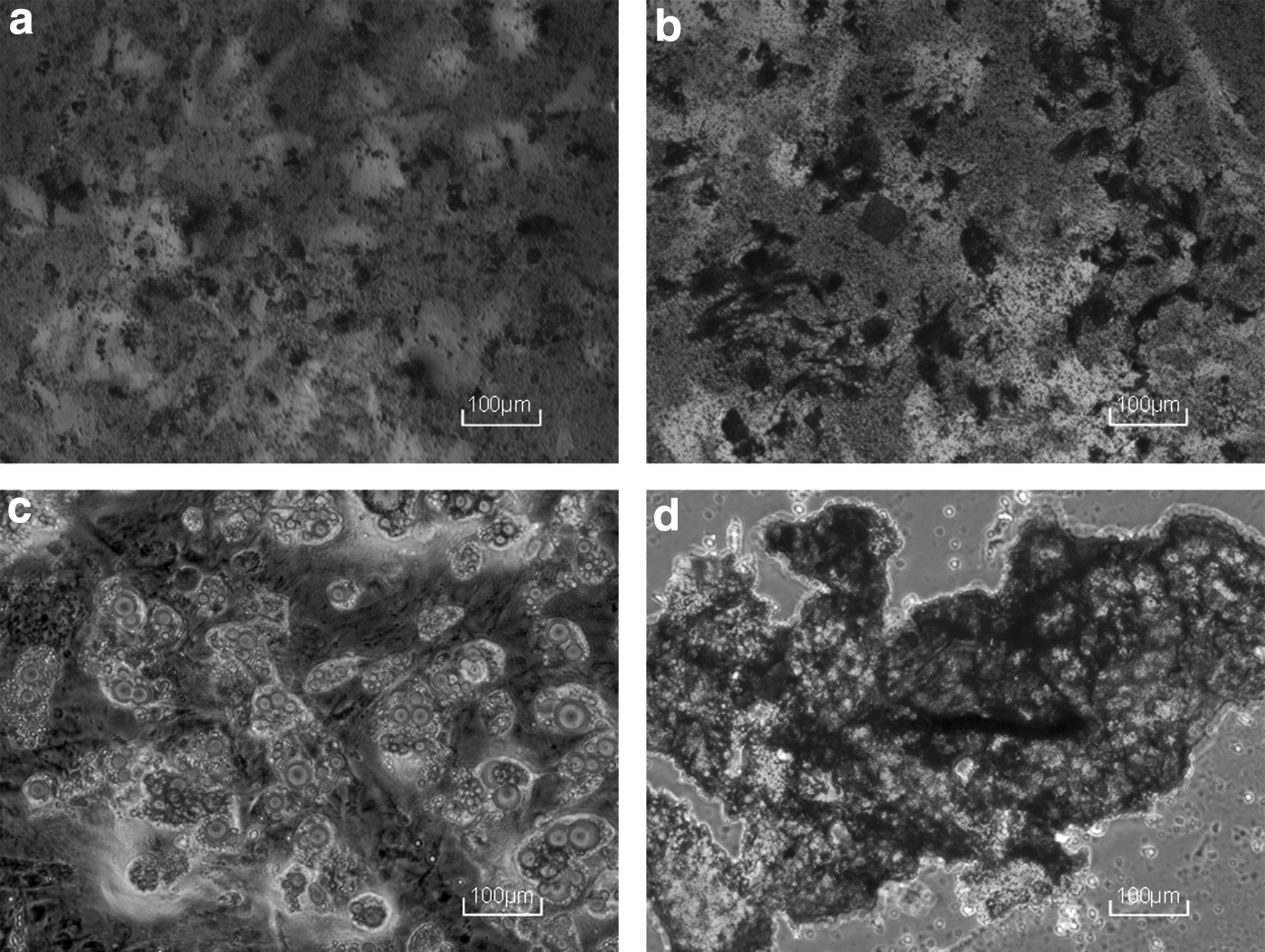
Differentiation of BMSCs.
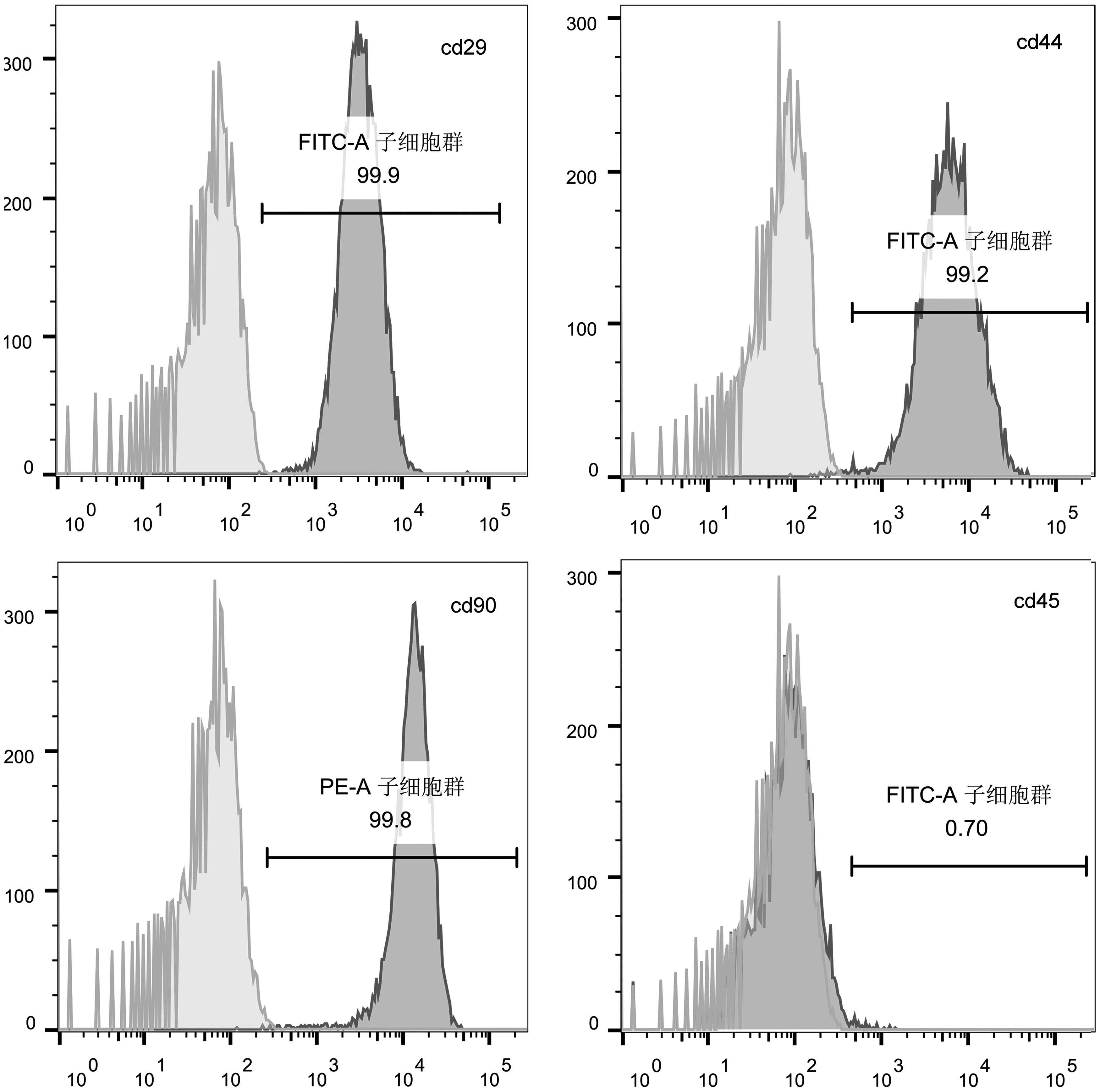
Positive expression rate of markers of BMSCs.
Basic properties of both scaffolds
White, porous scaffolds were produced by lyophilization and crosslinking. The C/G/A-dECM scaffolds exhibited open pores and thin walls on the surface (Fig. 2a, b). The interior was well interconnected (Fig. 2c, d). The scaffolds displayed an average pore size of 285.93 ± 85.39 μm, an average porosity of 90.62 ± 3.65%, an average water uptake ratio of 13.73 ± 1.16, and an average compressive modulus of 0.87 ± 0.05 kPa (Table 1).
The C/G scaffolds possessed a similar superficial and internal structure as the C/G/A-dECM scaffolds. The average pore size was 225.6 ± 45.71 μm; the average porosity was 88.47 ± 2.12%; the average water uptake ratio was 11.07 ± 0.87; and the average compressive modulus was 1.08 ± 0.07 kPa (Table 2).
C/G, chitosan/gelatin; C/G/A-dECM, chitosan/gelatin/adipose-derived extracellular matrix.
In vitro cytotoxicity
For the mixtures containing elute obtained at 1 and 3 days, the OD values of both scaffolds were similar to that of a medium without eluate. The OD value of the C/G/A-dECM eluate from 7 days was higher (p < 0.05) than the others, and the OD value of the C/G eluate from 7 days was similar to that of a medium without eluate (Fig. 5; p > 0.05).
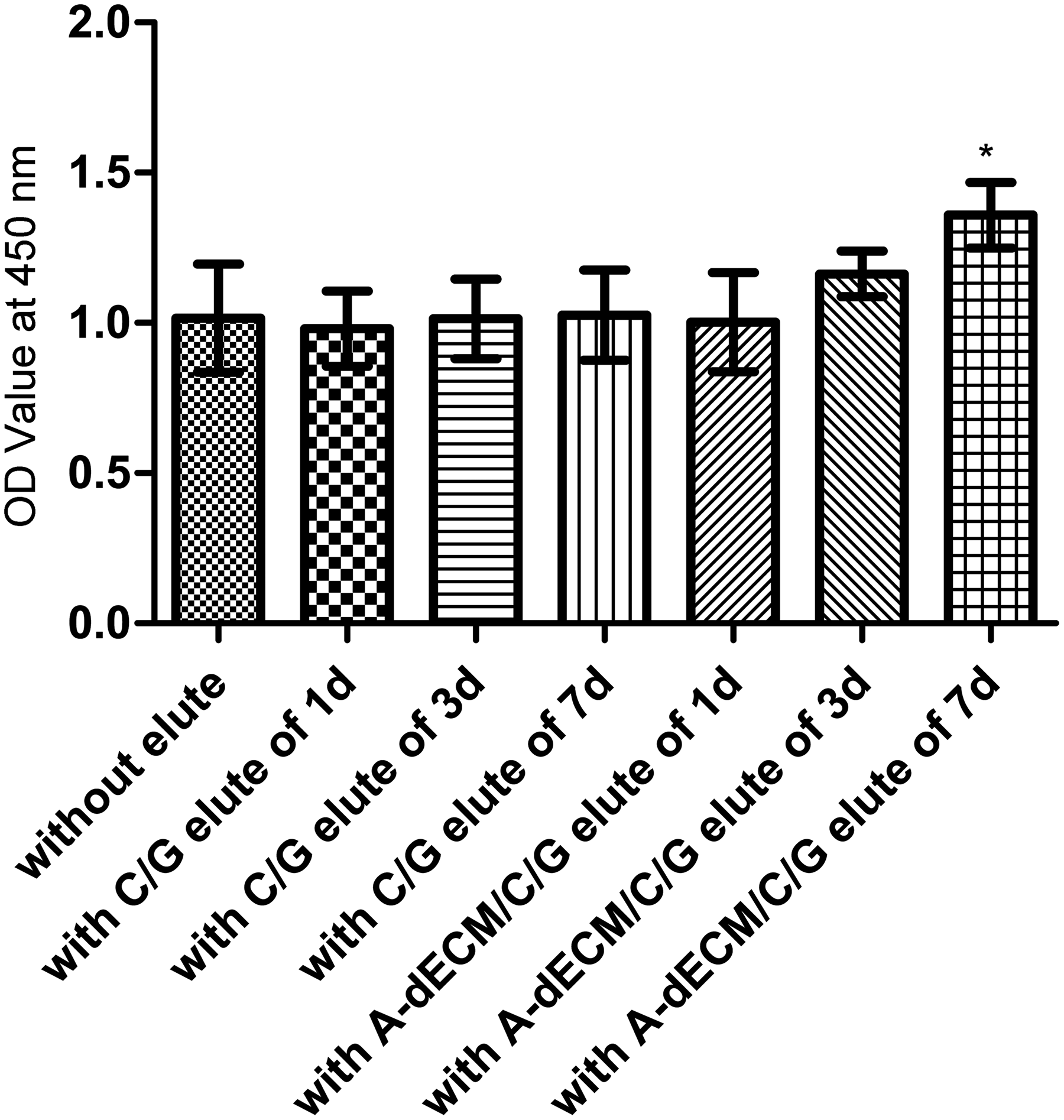
Cell proliferation after 3 days. *p < 0.05. OD, optical density.
Cell adhesion and cytoactivity
The average cell attachment ratio of the C/G/A-dECM scaffold was 51.25 ± 5.42%, which was higher than that of the C/G scaffold (39.17 ± 4.38%) (p < 0.05). Regarding fluorescence staining, when cells were cultured for 1 or 7 days, the cells on both scaffolds were almost uniformly green (Fig. 6). In the SEM analysis conducted at 7 days, the cells were observed to spread well on both scaffolds (Fig. 7), but especially on the C/G/A-dECM scaffold (Fig. 7c, d). ECM secreted by live cells on the C/G/A-dECM scaffold had covered the walls of the pores, and the cells had stretched well.

Dead/live stain for cells on scaffolds.

SEM of cell-seeded scaffolds.
Alkaline phosphatase
Figure 8 shows the ALP activity of BMSCs cultured on both scaffolds with or without OS reagents. In the presence of OS reagents, the U value of the C/G/A-dECM scaffold was higher than that of the C/G scaffold at 7 days (p < 0.01), 14 days (p < 0.01), and 21 days (p < 0.01). Without the OS reagent, the U value of the C/G/A-dECM scaffold was similar to that of the C/G scaffold at 7, 14, and 21 days.

U values of ALP after 7, 14, and 21 days. **p < 0.01. ALP, alkaline phosphatase.
Osteogenic markers
Figure 9 shows the expression of osteogenic markers. In the presence of the OS reagent, the ratios of opn mRNA, runx2 mRNA, alp mRNA, and ocn mRNA versus gapdh mRNA increased with time for both scaffolds. The levels were all higher for the C/GA-dECM scaffold than the C/G scaffold (p < 0.05 or 0.01).
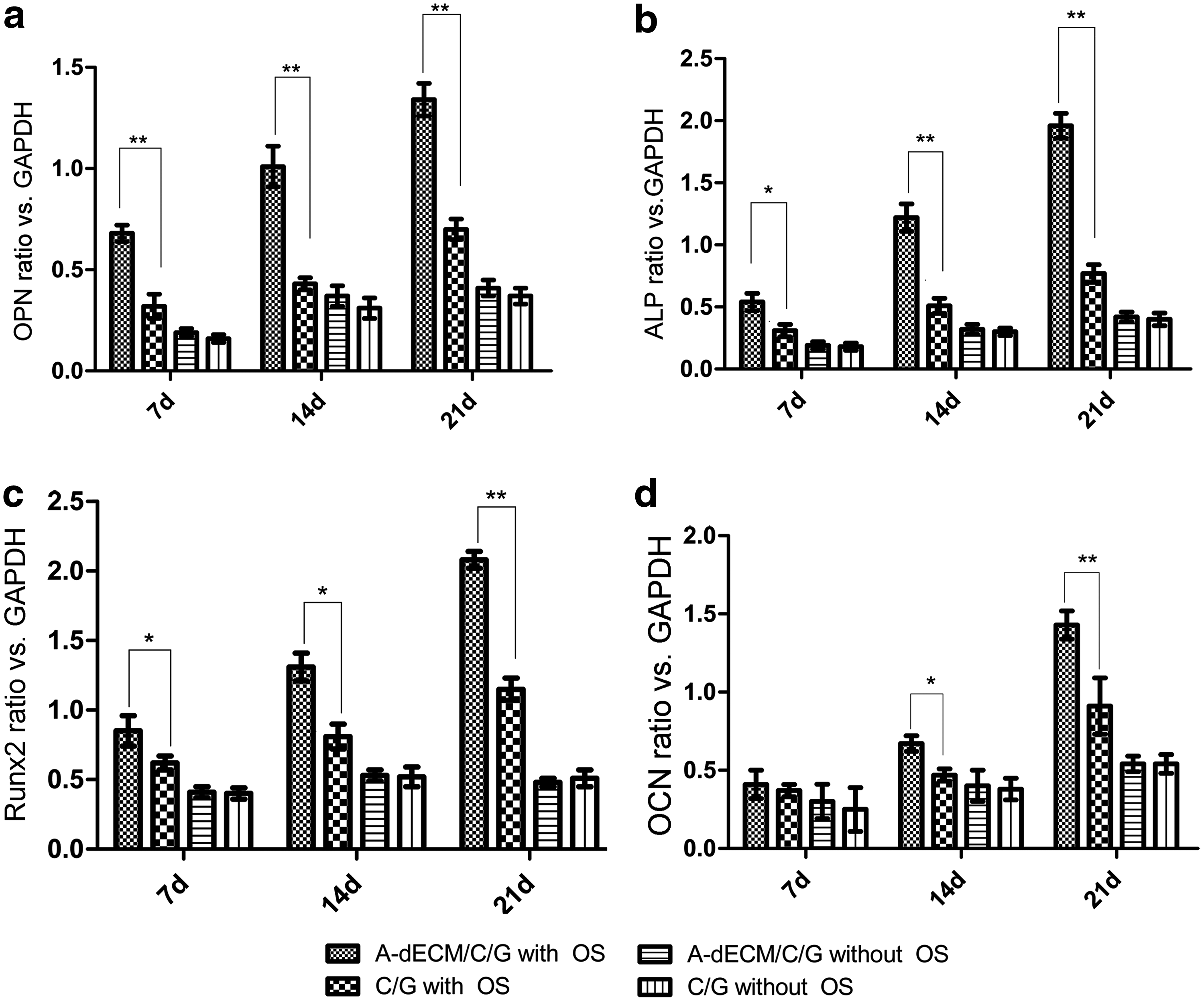
Osteogenic marker mRNA ratios versus GAPDH after 7, 14, and 21 days.
Without the OS reagent, the ratios were much lower than in the presence of the OS reagent, and increased slightly. The difference between the two scaffolds was insignificant (p > 0.05).
Mineralization deposition
Following alizarin red staining after 21 days of culture, the calcium nodules were stained fuchsia (Fig. 10). In the presence of the OS reagent, the nodules in the C/GA-dECM scaffold were larger and thicker than those in the C/G scaffold (Fig. 10a, b). Without the OS reagent, the nodules were very small and dispersed and were similar between the two scaffolds (Fig. 10c, d).

Alizarin red staining for calcium nodules.
Under von Kossa staining, the staining of the calcium nodules was similar to alizarin red staining (Fig. 11). However, the intensity of von Kossa staining appeared stronger than that of alizarin red staining.

Von Kossa for calcium nodules.
Discussion
ECM, which contains various growth factors and proteins, is increasingly applied to tissue-engineered scaffolds because of its bioactivity.4,25–27 However, suspensions of ECM exhibit bad stickiness; a portion of the ECM is therefore lost when lyophilized, and the resulting scaffolds are weak. Chitosan is an osteogenic material that is often used with gelatin.12,13,28 Scaffolds composed of C/G can provide proper strength and pore structure, providing a good microenvironment for cell attachment and proliferation. However, the osteogenic capacity of scaffolds does not satisfy researchers, and they therefore add various growth factors to different scaffolds via cell transfection to improve bioactivity for better osteogenesis.
In theory, the association of C/G with ECM can not only strengthen ECM scaffolds but also improve the bioactivity of the C/G scaffolds, without impairing their osteogenic character. To our knowledge, this type of scaffold has never been used for bone tissue engineering; thus, we examined whether A-dECM was beneficial to BMSCs and whether C/G/A-dECM scaffolds were a candidate for bone tissue engineering in this study.
In this study, we generated C/G/A-dECM scaffolds and C/G scaffolds. The appearance and interior structure of the two scaffolds were similar. The pore size of the C/G/A-dECM scaffolds was larger than that of the C/G scaffolds (p < 0.05). There was no significant difference between the two groups in terms of porosity (p > 0.05). The interior pore structure is beneficial for the provision of nutrient sustenance and exclusion of metabolic waste in cells, necessary for cell proliferation. 29 The ideal pore size is 100–350 μm, 30 and the two scaffolds exhibit a suitable pore size, which provides a good microenvironment for bone repair and regeneration. The C/G/A-dECM scaffolds displayed a better water uptake ratio than the C/G scaffolds. First, this is beneficial to the nutrient-providing and waste-excluding systems in in vitro culture and in vivo implantation. 31 Second, after taking up water, the scaffolds swell, which causes them to come into closer contact with the surrounding structure and avoids displacement of the implant. 32 The water uptake ratio of C/G/A-dECM was >13, which is beneficial for two reasons. First, the greater porosity increases contact between the medium and the pore wall. Second, hydrophilic groups in A-dECM, such as fibronectin, promote water absorption. 33
After replacing 1% C/G with 1% A-dECM, the compressive modules of the C/G/A-dECM scaffolds were less than those of the C/G scaffolds (p < 0.05). However, the compressive module was still much larger than that of the ECM scaffold reported previously. 4 Crosslinking strengthens the scaffolds, and the force between cationic chitosan and anionic gelatin is also beneficial. 12
When cells were cultured in the elutes of both scaffolds assessed in this study, the cell proliferation ratio did not decrease. Moreover, the cell proliferation ratio at 3 days in the presence of the 7-day eluate of the C/G/A-dECM scaffolds was higher than in the presence of the C/G scaffold eluate or no eluate. This result indicated that there was no residual glutaraldehyde in the scaffolds following reagent neutralization and ddw flushing and that the scaffolds did not inhibit cell growth. In addition, the C/G/A-dECM scaffolds can accelerate cell proliferation through probably slowly releasing bioactive factors into the medium, which is needed to be further studied.
Both scaffolds exhibited a good cell attachment ratio because of their high porosity, allowing the cells to contact the pore walls. The ratio for the C/G/A-dECM scaffolds was higher than for the C/G scaffolds. Various gelatins and proteins can provide a microenvironment for cell attachment, and growth factors within A-dECM, such as TGF-β1, which can recruit cells in situ, 34 may also promote attachment. When cultured for 1 or 7 days, the cells in the C/G/A-dECM scaffolds were more active than those in the C/G scaffolds according to the results of fluorescence staining and SEM analysis.
Osteoblast differentiation of BMSCs is the key process in bone formation. For the C/G/A-dECM scaffolds, ALP reached a high level following culture for 7 days with OS and continued to increase with time. ALP in the C/G scaffolds exhibited a similar trend, with a low U value (p < 0.01). Without OS, the U value of ALP was low, and there was no significant difference between the two scaffolds (p > 0.05), indicating that the scaffolds themselves display osteogenic ability. We speculate that although there were more cells in the C/G/A-dECM scaffolds than the C/G scaffolds, the greater amount of chitosan in the C/G scaffolds can balance the osteogenic capability. As markers of osteoblast differentiation, we chose to assess the relative gene expression ratios of opn, runx2, alp, and ocn versus gapdh. In the presence of the OS reagent, the ratios were significantly different (p < 0.01 or 0.05) in the C/G/A-dECM scaffolds and the C/G scaffolds, and they increased with time for both scaffolds. Without the OS reagent, there was no significant difference between the two scaffolds.
To observe mineralization, alizarin red staining and von Kossa staining were conducted. Both alizarin red and von Kossa stainings showed that in the presence of the OS reagent, the calcium nodules in the C/G/A-dECM scaffold were larger and more intensive than in the C/G scaffold. Moreover, the intensity of von Kossa staining appeared greater than that of alizarin red staining. We speculate that the use of different staining methods and staining times resulted in the different staining intensities of the calcium nodules. Without the OS reagent, alizarin red and von Kossa staining showed no difference between the calcium nodules in the C/G/A-dECM scaffolds and the C/G scaffolds, and the nodules were few in number and scattered. However, because of the multilayered structure of the scaffolds, we could not quantify the calcium nodules. Taking the results for ALP, mRNA of markers, and calcium deposition together, we conclude that C/G/A-dECM scaffolds can potentially be beneficial for osteoblast differentiation with an OS reagent.
A dynamic attachment method, rather than dropping a cell suspension onto scaffolds, was applied in this study. Considering the process of osteogenic differentiation of BMSCs, briefly, BMSCs are mobilized into the peripheral blood and transferred to an appropriate position for osteogenesis due to in situ osteogenic factors and the microenvironment. We are of the opinion that this dynamic method can simulate the process of the osteogenic differentiation of BMSCs, although later may lead to a higher attachment ratio.
There are some limitations to our study. First, decellularized adipose tissue may contain components other than A-dECM; thus, purification of the A-dECM is needed. Second, there are differences between in vivo and in vitro environments, and additional studies will therefore be required to assess the in vivo utility of the A-dECM scaffolds.
Conclusions
In summary, this study demonstrated that A-dECM can promote the attachment and proliferation of BMSCs, and C/G/A-ECM scaffolds are a candidate for bone tissue engineering.
Footnotes
Acknowledgment
This research was supported by the National Natural Science Foundation of China (Grant No. 304000573).
Disclosure Statement
No competing financial interests exist.