
Abstract
Dental pulp stem cells (DPSCs) are an attractive cell source for use in cell-based therapy, regenerative medicine, and tissue engineering because DPSCs have a high cell proliferation ability and multidifferentiation capacity. However, several problems are associated with the collection and preservation of DPSCs for use in future cell-based therapy. In particular, the isolation of DPSCs for cryopreservation is time consuming and expensive. In this study, we developed a novel cryopreservation method (NCM) for dental pulp tissues to isolate suitable DPSCs after thawing cryopreserved tissue. Using the NCM, dental pulp tissues were cultured on adhesion culture dishes for 5 days and then cryopreserved. After thawing, the cryopreserved dental pulp tissue fragments exhibited cell migration. We evaluated each property of DPSCs isolated using the NCM (DPSCs-NCM) and the explant method alone without cryopreservation (DPSCs-C). DPSCs-NCM had the same proliferation capacity as DPSCs-C. Flow cytometry (FACS) analysis indicated that both DPSCs-NCM and DPSCs-C were positive for mesenchymal stem cell markers at the same level but negative for hematopoietic cell markers. Moreover, both DPSCs-NCM and DPSCs-C could differentiate into osteogenic, chondrogenic, and adipogenic cells during culture in each induction medium. These results suggest that DPSCs-NCM may be mesenchymal stem cells. Therefore, our novel method might facilitate the less expensive cryopreservation of DPSCs, thereby providing suitable DPSCs for use in patients in future cell-based therapies.
Introduction
R
Gronthos et al. showed that MSCs may be present in human dental pulp tissues,15,16 which are referred to as dental pulp stem cells (DPSCs). DPSCs have a higher cell proliferation ability than other stem cell types and their differentiation capacity is similar to that of BMSCs and AMSCs.17,18 In addition, DPSCs are easy to collect without morbidity in the donor site because they are obtained from extracted teeth as medical waste. Recently, many researchers have investigated the clinical application of DPSCs in cell-based therapy, particularly in the regeneration of alveolar bone and dental pulp tissue. Furthermore, it has been suggested that DPSCs may be efficacious for the treatment of spinal cord injury, Alzheimer's disease, and Parkinson's disease based on animal studies.19–22 Therefore, DPSCs might be more feasible as a cell source for use in cell-based therapy.
The effective isolation of DPSCs is an important factor that affects the use of DPSCs in future cell-based therapy. In the current method used for the cryopreservation of DPSCs, dental pulp tissues are first extracted from the teeth, before DPSCs are isolated and cultured until the number of cells is sufficient, and they are then cryopreserved. Therefore, a long culture period is required from cell isolation until cryopreservation. In addition, this long-term culture period incurs excessive labor costs as well as other expenses, and there is a risk of potential contamination by microorganisms. Thus, a safe, easy, and inexpensive approach is needed for banking DPSCs. In this study, we developed a novel cryopreservation method (NCM) to facilitate the more effective collection of human DPSCs. In particular, we examined how dental pulp tissues must be cryopreserved to collect cells effectively from tissues and we investigated the characteristics of the cells recovered from tissues using the novel cryopreservation system, including the expression of MSC markers, cell growth ability, and differentiation capacity.
Materials and Methods
Collection of dental pulp tissues
The dental pulp tissues were obtained from the third molars of healthy patients (18–34 years of age) extracted at Tsurumi University Dental Hospital. Before extraction, we obtained informed written consent from all patients. This study was approved by the Research Ethics Review Committee at Tsurumi University School of Dental Medicine (approval number: 901). The third molars used in this study had no caries, restoration, or inflamed pulp tissue. The extracted teeth were cut at the cement–enamel junction using a diamond bur and the dental pulp tissues were collected gently under sterile conditions.
Histological examination of dental pulp tissue cultured using the explant method
First, we histologically observed the process from the start of culture until cell migration from the pulp tissue pieces using the explant method, which is the conventional technique employed for isolating stem cells. The dental pulp tissues in the crown part were divided into two parts. One fragment was placed onto a 60-mm culture dish (Falcon Corning, New York) and cultured in α-modified Eagle's medium (α-MEM; Sigma-Aldrich, St Louis) supplemented with 10% fetal bovine serum (FBS; Biological Industries, Kibbutz Beit-Haemek, Israel), 100 U/mL penicillin, 100 μg/mL streptomycin (Invitrogen, Carlsbad, CA), and 1× Glutamax (Invitrogen), for 5 days at 37°C and under 5% CO2. These fragments were then fixed with 4% paraformaldehyde phosphate buffer solution (PFA; Wako Pure Chemical Industries Ltd., Osaka, Japan), embedded in paraffin, and cut to a thickness of 4 μm. The other fragments were not cultured and they were embedded immediately in paraffin. The paraffin sections were deparaffinized, hydrated, and stained with hematoxylin and eosin before making histological observations.
Immunohistochemistry was performed to examine the behavior of the cells within tissue fragments during explant culture. The sections were deparaffinized, hydrated, washed in phosphate-buffered saline (PBS, pH 7.4), and blocked by treating with 3% H2O2 in methanol for 1 h at room temperature and then with 10% normal goat serum at room temperature for 10 min. Next, the sections were incubated overnight at 4°C with rabbit anti-Ki-67 antibody (Abcam, Cambridge, United Kingdom) diluted to 1:250. After washing with PBS, the localization of Ki-67 was observed using a Histofine SAB-PO (R) kit (Nichirei Biosciences, Inc., Tokyo, Japan) and a 3,3′-diaminobenzidine (DAB) substrate kit (Nichirei Biosciences, Inc.). The sections were stained with DAB solution for 5 min and counterstained with hematoxylin. Negative control sections were run without the primary antibody. The other paraffin sections were deparaffinized, hydrated, washed in PBS, and stained using a TACS® two-terminal deoxynucleotidyl transferase-DAB in situ apoptosis detection kit (Trevigen). The sections were observed by microscopy.
NCM for dental pulp tissues
In the NCM, the fragments of dental pulp tissues were placed onto a 60-mm culture dish (Falcon Corning) and cultured in α-MEM (Sigma-Aldrich) supplemented with 10% FBS (Biological Industries), 100 U/mL penicillin, 100 μg/mL streptomycin (Invitrogen, Carlsbad), and 1× Glutamax (Invitrogen) for 5 days at 37°C and under 5% CO2, before storing overnight in a Cellbanker® 2 (Takara Bio, Inc., Shiga, Japan) at −80°C, and then freezing in liquid nitrogen for 1 week.
In the control group, dental pulp tissue fragments without culture were immediately cryopreserved (IC). The dental pulp tissues obtained from 10 subjects were used. The dental pulp tissues from one subject were separated into six fragments, which were then divided into two groups: (1) three fragments were cryopreserved by NCM and (2) the other three fragments by IC. After cryopreservation, these tissues were thawed quickly at 37°C and placed on a 60-mm culture dish (Falcon Corning) in α-MEM (Sigma-Aldrich) supplemented with 10% FBS (Biological Industries), 100 U/mL penicillin, 100 μg/mL streptomycin (Invitrogen, Carlsbad), and 1× Glutamax (Invitrogen) at 37°C and under 5% CO2. After culture for 10 days, we performed cell isolation evaluations using each tissue in the two groups.
Evaluations of cell characteristics
The whole coronal dental pulp tissue from each subject was divided into six fragments. Three fragments were cryopreserved by NCM. One week later, these fragments were thawed and placed in six-well plates, and the cells recovered from each fragment were cultured until semiconfluent and then transferred to 100 mm dishes, where they were allowed to expand continuously until passage 3 for use in further experiments. We designated these cells as DPSCs isolated by NCM (DPSCs-NCM) (Fig. 4). The other three fragments were used directly for cell isolation without cryopreservation, where the cells were expanded until passage 3, as already described, and designated as control DPSCs (DPSCs-C) (Fig. 4).
Cell proliferation
Both the DPSCs-NCM and DPSCs-C were seeded at a density of 5 × 103 cells/well in 24-well plates (Falcon Corning) and cultured until each specified time point. The cells were fixed with 1% glutaraldehyde (Wako) and stained with 0.3% crystal violet (Sigma-Aldrich) at time points after 0, 1, 3, 5, 7, 9, and 12 days of culture. The absorbance of an extract obtained from each sample using 100% ethanol was measured in triplicate at 570 nm using a microplate reader (Bio-Rad, California).
Flow cytometry analysis
DPSCs-NCM and DPSCs-C were cultured in 75 cm2 cell culture flasks (Falcon Corning) until 60% confluence. The cells were resuspended in flow cytometry staining buffer (PBS + 2% FBS) at a concentration of 1 × 106 cells/tube, stained with primary specific antibodies, that is, PE-conjugated CD146, allophycocyanin (APC)-conjugated CD73, APC-conjugated CD105, APC-conjugated CD34, APC-conjugated CD45, APC-conjugated CD14, and APC-conjugated HLA-DR (eBioscience, San Diego), and incubated for 30 min at 4°C. Resuspended cells were stained with Stro-1 primary antibody (Abcam) and incubated for 30 min at 4°C, followed by the secondary antibody (FITC-conjugated: Abcam) for 30 min. Flow cytometry was performed with a FACSCalibur flow cytometer (BD Bioscience, San Jose). The number of positive cells was determined using FACSDiva software (BD Bioscience).
Osteogenic differentiation
DPSCs-NCM and DPSCs-C were seeded in six-well plates (Falcon Corning), cultured until confluent in maintenance medium, which comprised α-MEM with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin, and then induced with osteogenic medium comprising α-MEM with 100 nM dexamethasone (Sigma-Aldrich), 2 mM β-glycerophosphate (Sigma-Aldrich), and 50 μM ascorbic acid 2-phospate (Wako). 23 As controls, DPSCs-NCM and DPSCs-C were cultured only in the maintenance medium. The medium was changed twice per week. After induction for 21 days, the cells were fixed in 4% PFA. The cells were then stained with 1% Alizarin red-S (Sigma-Aldrich) for 20 min at room temperature. The Alizarin red-positive area was analyzed using Image-J (National Institute of Health) and expressed as the percentage of the Alizarin red-positive area relative to the total area. 24 Moreover, the mRNA expression levels of osteogenic-specific genes, that is, osteocalcin (OCN), Runt-related gene 2 (RUNX2), dentin sialophosphoprotein (DSPP), and alkaline phosphatase (ALP), were analyzed by semiquantitative reverse transcription polymerase chain reaction (RT-PCR).
Adipogenic differentiation
DPSCs-NCM and DPSCs-C were seeded in six-well plates, cultured until confluent in maintenance medium, and then induced with adipogenic medium, which comprised 1 μM dexamethasone (Sigma-Aldrich), 100 μM indomethacin (Wako), 500 μM 3-isobutyl-l-methylxanthine (Wako), and 10 μg/mL human insulin (Wako). 23 As controls, DPSCs-NCM and DPSCs-C were cultured in the maintenance medium alone. The medium was changed twice per week. After induction for 21 days, the cells were fixed in 4% PFA and stained with 0.2% Oil Red O (Sigma-Aldrich) solution to detect lipid droplets. To determine the amount of lipid droplets, Oil Red O stain was extracted from each well with 60% isopropanol and the absorbance of the extracted stain was then measured at 510 nm using a spectrophotometer (Bio-Rad). Moreover, the mRNA expression levels of adipocyte-specific genes, that is, lipoprotein lipase (LPL) and peroxisome proliferator-activated receptor γ2 (PPARγ2), were analyzed by semiquantitative RT-PCR.
Chondrogenic differentiation
DPSCs-NCM and DPSCs-C were resuspended at a concentration of 2 × 105 cells in 1.0 mL of Dulbecco's modified Eagle's medium (Sigma-Aldrich) containing 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin before transferring to a 15-mL tube (Falcon Corning), centrifuging at 1000 rpm (120 ×g) for 5 min, and incubating at 37°C under 5% CO2. After culture for 2 days, the cells were formed into a pellet shape and induced with chondrogenic medium, which comprised 2 mM β-glycerophosphate (Sigma-Aldrich), 100 μM l-ascorbate 2 phosphate (Wako), 100 nM dexamethasone (Sigma-Aldrich), 10 ng/mL transforming growth factor beta-3 (TGFβ-3; R&D system, Inc., Minneapolis), 2 mM
Semiquantitative RT-PCR
Semiquantitative RT-PCR was performed to examine the mRNA expression levels of osteogenic-, adipogenic-, and chondrogenic-specific genes. The primer sequences and PCR amplification conditions are shown in Table 1. Total RNA was extracted from the cells using TRIzol reagent (Invitrogen) and cDNA was generated from 1 μg of the total RNA using the SuperScript III First-Strand Synthesis System (Invitrogen). PCR amplification was conducted in a 20 μL reaction mixture using 1.1 × ReddyMix PCR Master Mix (ABgene, Thermo Scientific, Waltham). The GAPDH gene was used as an internal quantitative and quality control for the cDNA. The PCR products were analyzed by ethidium bromide staining after separation using electrophoresis on a 2% agarose gel.
Statistical analysis
The results were represented as the mean ± standard errors. All data were representative of at least three individual experiments and were analyzed with Scheffé's test. Differences were considered significant at p < 0.05.
Results
Histological analysis of dental pulp tissue cultured using the explant method
The histological findings characterized the cell behavior within the dental pulp tissues during the explant process. Before culture, the cells were distributed heterogeneously within the tissue fragments (Fig. 1A). After culture for 5 days, many cells were present at the edges of the dish containing tissue fragments, whereas few cells were present in the center of the tissue fragments. Next, we used immunohistochemistry to investigate the detailed behavior of cells within the tissue fragments during the process. The results indicated that many cells at the edge of the tissue fragments did not express Ki-67, which is a marker of cell proliferation (Supplementary Fig. S1; Supplementary Data are available online at

Histological findings for dental pulp tissue fragments before and after explant culture. Before culture is indicated as 0 day for explant culture when the cells were distributed heterogeneously within the tissue fragments. After explant culture for 5 days, many cells were present on the edges at the side of the dish containing the tissue fragments. The bottom part of the tissue was at the side of the dish. Magnified views are indicated as square areas at the left-hand side of the histological findings.
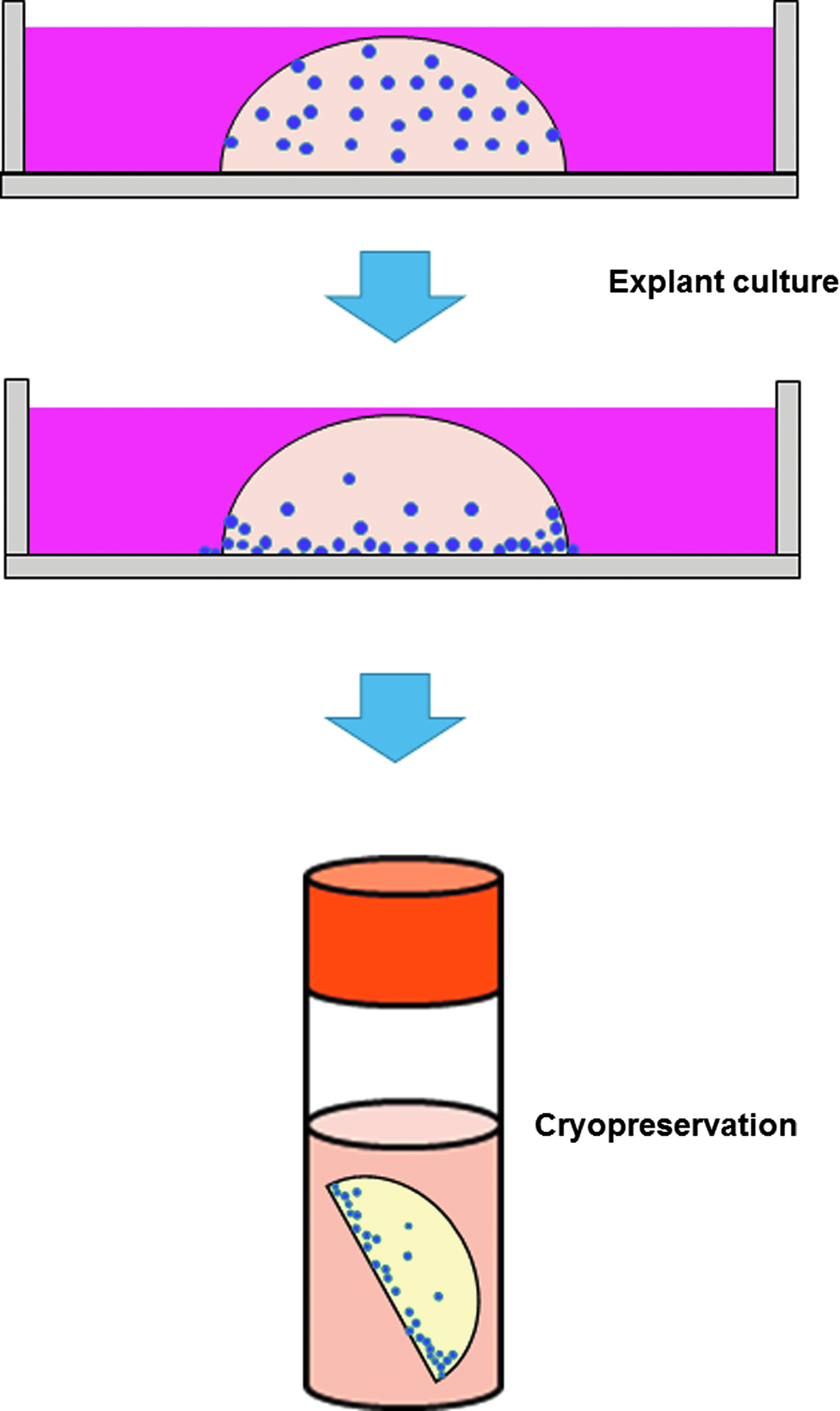
Scheme of the novel cryopreservation method (NCM). First, dental pulp tissue fragments were cultured for 5 days according to the explant method. Next, the fragments were cryopreserved in a Cellbanker 2.
Comparison of the isolation rate using IC and NCM
First, we evaluated whether cells were recovered from the tissues cryopreserved by NCM and IC (Fig. 3a). With NCM, 28 of the 30 tissue fragments exhibited cell isolation (93.3%) (Fig. 3b). By contrast, only 11 of the 30 IC fragments exhibited cell isolation (36.7%) (Fig. 3b). These results demonstrate that NCM could allow more cells to be recovered from cryopreserved tissues after thawing than IC.

Comparison of the outgrowth rates using the immediate cryopreservation (IC) method and NCM.
Proliferation assay
Next, we investigated the proliferation characteristics of DPSCs-NCM and DPSCs-C (Fig. 4). Phase-contrast microscopy showed that the DPSCs-NCM appeared to be fibroblast-like cells in the same manner as DPSCs-C (Fig. 5A). The number of cells of both DPSCs-NCM and DPSCs-C increased gradually during culture for 10 days and then stabilized. The crystal violet assay was performed to estimate the proliferation capacity of DPSCs-NCM and DPSCs-C. The proliferation capacity of DPSCs-NCM was slightly lower than that of DPSCs-C, but the cell proliferation rate did not differ significantly between DPSCs-NCM and DPSCs-C (Fig. 5B).

Scheme of the experiment used to compare the characteristics of DPSCs-NCM and DPSCs-C. As already described, the dental pulp tissue was separated into six fragments, which were then divided into two groups: DPSCs-C and DPSCs-NCM. DPSCs-C were isolated by explant culture and used without cryopreservation in the analysis. DPSCs-NCM were recovered from the tissues cryopreserved by the NCM. Finally, DPSCs-NCM and DPSCs-C were analyzed to determine the cell proliferation ability, expression of the immunophenotype, and the differentiation potential. DPSCs, dental pulp stem cells.
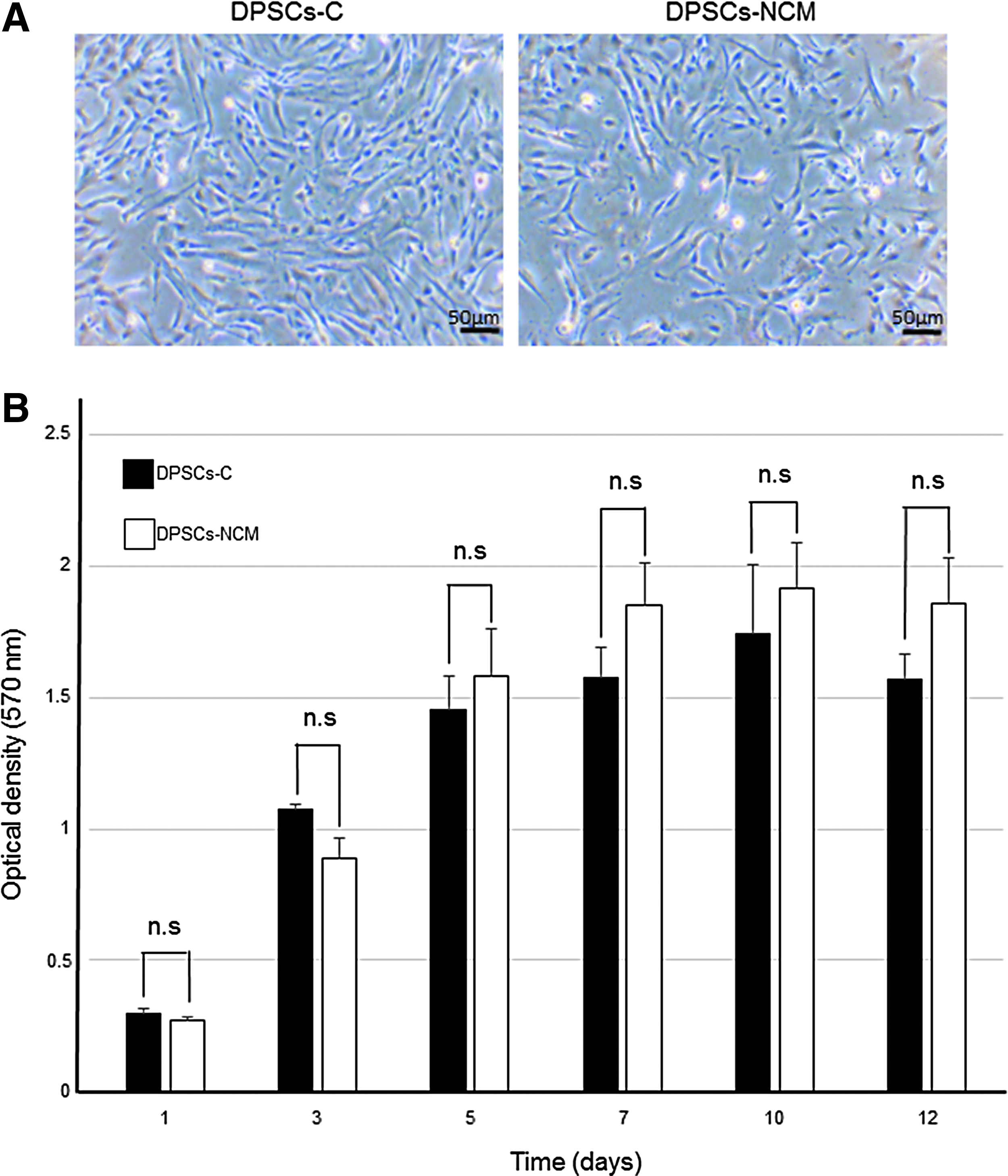
Morphological characteristics of DPSCs-NCM and DPSCs-C.
Flow cytometry analysis
We performed flow cytometry to assess the expression profiles of cell surface MSC markers in both DPSCs-NCM and DPSCs-C. The results demonstrated that both DPSCs-NCM and DPSCs-C were positive for Stro-1, CD146, CD73, and CD105 as MSC markers, but negative for CD34, CD45, CD14, and HLA-DR as hematopoietic cell markers. Moreover, the expression level of each marker in DPSCs-NCM was similar to that in DPSCs-C (Fig. 6). These results demonstrate that DPSCs-NCM had the immunophenotype of MSCs.

Representative histograms for cell surface markers in DPSC-NCM and DPSC-C. Each type of DPSC was analyzed at passage 3 using a flow cytometer (n = 3). The red line indicates the histogram for positive cells and the black line indicates the histogram for negative cells. Both DPSCs-NCM and DPSCs-C were positive for Stro-1, CD146, CD73, and CD105 as MSC markers, but negative for CD34, CD45, CD14, and HLA-DR as hematopoietic cells markers. MSC, mesenchymal stem cell.
Osteogenic differentiation
We used Alizarin Red S staining and semiquantitative RT-PCR to determine the osteogenic differentiation capacity of DPSCs-NCM. After osteogenic induction for 3 weeks, DPSCs-NCM and DPSCs-C formed mineralization nodules, which could be stained by Alizarin Red S. The Alizarin Red-positive area indicated that DPSCs-NCM formed a mineralization nodule with a size similar to that produced by DPSCs-C (Fig. 7A). Furthermore, RT-PCR analysis showed that DPSCs-NCM subjected to osteogenic induction expressed mRNAs for OCN, RUNX2, DSPP, and ALP, which are osteogenic markers (Fig. 7A). These results demonstrate that DPSCs-NCM had the capacity for osteogenic differentiation.

Multilineage differentiation capacities of DPSCs-DCM and DPSCs-C.
Adipogenic differentiation
We performed RT-PCR analysis and Oil Red O staining to confirm whether DPSCs-NCM could differentiate into adipocytes. DPSCs-NCM and DPSCs-C were cultured in the adipogenic differentiation medium for 3 weeks. After induction, DPSCs-NCM expressed mRNAs for LPL and PPARγ2 (Fig. 7B). In addition, the expression of these adipocyte-specific genes was confirmed by RT-PCR analysis (Fig. 7B). DPSCs-NCM formed Oil Red O-positive lipid droplets similar to those in DPSCs-C. Quantification using Oil Red O detected no significant differences between DPSCs-NCM and DPSCs-C. These results indicate that DPSCs-NCM had the capacity for adipogenic differentiation.
Chondrogenic differentiation
Finally, we evaluated whether DPSCs-NCM differentiated into chondrocytes. After chondrogenic induction for 3 weeks, both DPSCs-NCM and DPSCs-C formed pellets, whereas DPSCs-NCM and DPSCs-C without chondrogenic induction did not form pellets. The histological findings demonstrated that the cell pellets produced from DPSCs-NCM had an Alcian Blue-positive chondrogenic matrix, and immunohistochemically, they expressed AGG as a chondrogenic marker in the same manner as DPSCs-C (Fig. 7C). Furthermore, semiquantitative RT-PCR indicated that DPSCs-NCM expressed mRNAs for chondrogenic makers, that is, AGG, COL II, and SOX9 (Fig. 7C). These results indicate that DPSCs-NCM had the capacity for chondrogenic differentiation.
Discussion
In this study, we developed a novel cryopreservation system for dental pulp tissues to facilitate the efficient banking of DPSCs. We showed that cells could be recovered more effectively from tissues preserved using the novel cryopreservation system compared with those from IC tissues. In addition, the growth potential and differentiation abilities of the cells recovered from the cryopreserved tissue after thawing were similar to those of MSCs in the same manner as cells isolated from noncryopreserved dental pulp tissue, thereby suggesting that the novel cryopreservation system is feasible for banking DPSCs, and thus it could be used in future cell-based therapy.
Tissue engineering and regenerative medicine have developed as cell-based therapies using DPSCs, and many studies have focused on improving the effectiveness and safe storage of DPSCs. 26 In the current storage procedure (banking system), the process from cell isolation to cryopreservation requires about 3 weeks. Moreover, long-term culture increases the risk of contamination, as well as costs and labor requirements. Therefore, reducing the processing time would be highly advantageous. In fact, recent studies have tried to develop a banking system that does not require complex cell processing before cryopreservation. The IC tissue method is known to be the quickest and simplest storage method, and it has been applied to the acquisition of DPSCs. 27 Lindemann et al. indicated that the cell recovery rate from IC whole pulp tissues was very low at 30%. Tirino et al. also demonstrated that no stem cells could be obtained from IC dental pulp tissues despite conducting experiments to improve the collection rate for cells. 28 In this study, our results indicated that similar to previous studies, the successful isolation rate from IC tissue was also very low at 36.7%. In addition, the histological findings for IC dental pulp tissues after thawing showed that few viable cells remained within the tissues (data not shown). In general, a cryopreservation agent must be added to protect tissues or cells from damage because of the formation of ice crystals in the tissues. However, few viable cells could be recovered from IC dental pulp tissues despite the application of a cryopreservation agent. Based on our results and previous studies, this may be attributable to insufficient infiltration of the cryopreservation agent into the center of the tissues. 29 Therefore, adequate infiltration of the cryopreservation agent into cells in the tissue may be important for recovering more viable cells from cryopreserved tissue. Thus, we focused on creating an environment for tissues that allowed the cryopreservation agent to infiltrate more readily. In our system, explant culture for only 5 days before cryopreservation increased the successful isolation rate from cryopreserved tissue to nearly 90%, although the underlying explanation for this improvement remains unclear. However, histological findings based on explant culture showed that viable cells present in the borders of the tissues were more readily infiltrated by the cryopreservation agent. In addition, the cells recovered from the cryopreserved tissues retained a high successful isolation rate (Fig. 3). Therefore, these findings indicate that 5 days of culture before cryopreservation can produce cells with a high capacity for migration to the area where the cryopreservation agent is easily accessible. In the future, we will try to elucidate the details of this mechanism.
Currently, two methods are available for the isolation of DPSCs from dental pulp tissues: enzyme digestion and explant methods. 30 The enzyme digestion method requires collagenase and dispase, which are considered to damage cells. Recently, the Food and Drug Administration (FDA) has provided recommendations regarding the criteria for minimal manipulation (21 CFR Part 1271) to manufacturers of human cells, tissues, and cellular and tissue-based products. According to these recommendations, minimal manipulation is defined as processing that does not alter the relevant biological characteristics of cells or tissues. Therefore, the FDA has also stated that enzymatic digestion does not meet the regulatory definition of minimal manipulation. By contrast, the explant method satisfies the definition because there is no requirement for enzyme treatment before isolation. This is why we selected the explant method for isolating DPSCs from cryopreserved tissues. DPSCs isolated using the explant method (DPSCs-C) had characteristics, immunophenotypes, and capacities for proliferation and differentiation very similar to DPSCs treated by enzyme digestion (DPSCs-E). We found that the colony-forming ability and Stro-1 expression level were higher in DPSCs-E than in DPSCs-C, but the in vivo hard tissue-forming ability was superior in DPSCs-C than in DPSCs-E. 23 In the future, we will perform rigorous comparisons of DPSCs-NCM and DPSCs-E.
The characteristics of DPSCs-NCM were similar to those of DPSCs-C, which were isolated from noncryopreserved tissues (Figs. 5–7). Moreover, the DPSCs-NCM met the criteria for MSCs. The International Society for Cellular Therapy defined MSCs based on three criteria: (1) MSCs must be plastic adherent when maintained in standard culture conditions; (2) MSCs must be positive for CD105, CD146, CD73, and Stro-1 surface antigens, but negative for HLA-DR, CD11b, CD14, CD34, CD45, and CD31; and (3) MSCs must differentiate in vitro into osteocytes, chondrocytes, and adipocytes. 31 In this study, DPSCs-NCM expressed the MSC markers but not the hematopoietic cell markers, and they were able to differentiate into osteoblasts, chondrocytes, and adipocytes under each of the specific culture conditions (Figs. 6 and 7). Therefore, our results confirmed that DPSCs-NCM had the same characteristics as MSCs.
It is necessary to obtain sufficient amounts (clinical scale number) of DPSCs for their future clinical applications. 32 In this study, the growth rate of DPSCs-NCM was slightly lower than that of DPSCs-C, but the cell growth capacity of DPSCs-NCM was sufficient after three passages (Fig. 5B).
Recent studies have investigated the effects of xenogenic-free products and serum-free media on the isolation and culture of DPSCs, thereby demonstrating that they do not alter the behavior of DPSCs, cell growth capacity, or differentiation ability.33,34 In this study, we did not use these products but we consider that these products might not affect our system, as shown in previous studies. In the future, we will confirm whether xenogenic-free products affect our cryopreservation system.
In this study, we developed a simple and efficient cryopreservation system for collecting dental pulp tissues. This system should be improved before its clinical use. Moreover, we plan to produce a supplementary device to simplify the process from the collection of dental pulp tissues to cryopreservation using our system. In particular, the device should be able to culture dental pulp tissues during their transportation from a clinic to a laboratory, so they can be IC upon their arrival. In future research, we will improve the feasibility of our system for banking DPSCs in clinical applications.
Footnotes
Acknowledgments
This study was supported by the Division of Pathology, Department of Oral Diagnostic Sciences, School of Dentistry, Showa University, and a Grant-in-Aid for Scientific Research (23792392) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.