
Abstract
A common challenge in cell therapy is the inability to routinely maintain survival and localization of injected therapeutic cells. Delivering cells by direct injection increases the flexibility of clinical applications, but may cause low cell viability and retention rates due to the high shear forces in the needle and mechanical wash out. In this study, we encapsulated endothelial colony forming cells (ECFCs) in poly(ethylene glycol)-fibrinogen (PF) hydrogel microspheres using a custom-built microfluidic device; this system supports rapid encapsulation of high cell concentrations (10 million cells per mL) and resulting cell-laden microspheres are highly uniform in shape and size. The encapsulated ECFCs were shown to have >95% viability and continued to rapidly proliferate. Expression of cell markers (von Willebrand factor, CD105, and CD14), the ability to form tubules on basement membrane matrix, and the ability to take up low-density lipoprotein were similar between pre- and post-encapsulated cells. Viability of encapsulated ECFCs was maintained after shear through 18–23-gauge needles. Ex vivo and in vivo cell delivery studies were performed by encapsulating and injecting autologous equine ECFCs subcutaneously into distal limb full-thickness wounds of adult horses. Injected ECFCs were visualized by labeling with fluorescent nanodots before encapsulation. One week after injection, confocal microscopy analysis of biopsies of the leading edges of the wounds showed that the encapsulated ECFCs migrated into the surrounding host tissue indicating successful retention and survival of the delivered ECFCs. Rapid, scalable cell encapsulation into PF microspheres was demonstrated to be practical for use in large animal cell therapy and is a clinically relevant method to maintain cell retention and survival after local injection.
Introduction
T
Encapsulation of stem or progenitor cells in hydrogels has been shown to support cell proliferation and long-term survival. 15 In addition, hydrogels can act as semipermeable media to protect the transplanted or delivered cells from the host immune system. 16 Cell retention at the desired location can be significantly improved by encapsulating cells in a hydrogel matrix before delivery. 17 Therefore, cell encapsulation in hydrogel scaffolds could advance the potential of cell-based therapies.
The natural/synthetic hybrid hydrogel poly(ethylene glycol) (PEG)-fibrinogen (PF) has been shown to support a range of tissue engineering applications,18–22 including angiogenesis. While the acrylated-PEG enables rapid formation of a supportive structure through photocrosslinking, the fibrinogen provides adhesive anchorage and degradability for cellular activity. A wide variety of cell types, including smooth muscle cells, induced pluripotent stem cells, and chondrocytes, have been encapsulated in PF with minimal impact in cell viability.23–27 Furthermore, injection of cells encapsulated within PF has been shown to enhance cell survival and differentiation compared to injection of cells suspended in aqueous saline solution, 28 making PF a suitable biomaterial for cell delivery.
Encapsulation of cells into hydrogel microspheres increases the flexibility of use of resulting engineered tissues for clinical applications, because of the ability to deliver microspheres by injection. However, typical cell encapsulation in microspheres using microfluidic devices is limited by low cell density, which makes delivery of sufficient cell numbers in reasonable volumes for therapeutic applications challenging. 16
In this study, we present encapsulation of ECFCs in PF hydrogel microspheres at a high concentration of 10 million cells per mL using a custom-built microfluidic device. The microspheres are highly uniform in shape and size. The encapsulated ECFCs were shown to have high viability post-encapsulation and their phenotype was preserved compared to cells in typical cell culture conditions. As an initial proof of concept for utilizing microspheres produced by the microfluidic device as clinical cell delivery vehicles, an in vivo cell delivery study was also performed by encapsulating and injecting autologous equine ECFCs into a distal limb wound model in adult horses. The outcomes of this study demonstrate the capabilities of this cell encapsulation platform and examine its potential for supporting therapeutic cell delivery by injection.
Methods
Equine cell isolation and culture
All procedures involving animals were approved by the Auburn University Animal Care and Use Committee. Isolation and culture of equine ECFCs from equine peripheral blood were performed based on a previously published method. 29 ECFCs were cultured in Endothelial Cell Basal Medium-2 (Lonza) containing 10% horse serum (HyClone) and SingleQuots Kit (Lonza) at 37°C and 5% CO2. The SingleQuots Kit contained hydrocortisone, hFGF-β, VEGF, R3-IGF-1, ascorbic acid, hEGF, GA-1000, and heparin.
The ECFCs were seeded and expanded on collagen-coated tissue culture polystyrene (TCPS) flasks. When ECFCs reached 90% confluency, cells were subcultured using trypsin/EDTA (Lonza) to detach the cells at 37°C for 50 s, followed by neutralization with fresh medium and centrifugation at 200 g for 5 min. ECFCs were resuspended in medium and then subcultured at a ratio of 1:6 or immediately used for experiments. Cells between passage 2–7 were used for all experiments.
PEG-diacrylate synthesis
Poly(ethylene glycol) (PEG, 10 kDa; Sigma) was acrylated to form PEG-diacrylate (PEGDA) following a previously published method. 30 Briefly, PEG was first lyophilized and then reacted with 0.4 M acryloyl chloride (Alfa Aesar) and 0.2 M triethylamine (TEA, Sigma) in anhydrous dichloromethane (Acros) under argon overnight. 1.5 M K2CO3 (Fisher) was added, and then the solution was separated into aqueous and organic phases. The organic phase was collected and dried with anhydrous MgSO4 (Fisher). The PEGDA was then precipitated by cold ethyl ether, filtered, dried, and stored under argon at −20°C. The degree of acrylation was estimated to be 96.0% by nuclear magnetic resonance.
PEG-fibrinogen synthesis
PF was synthesized by following a previously published method. 26 In brief, fibrinogen (Type I-S; Sigma) was dissolved in 8 M urea (Sigma) in phosphate-buffered saline (PBS) (Lonza) solution to a final concentration of 7 mg/mL with pH of 7.4. Tris (2-carboxyethyl) phosphine (Acros Organics) was added to the solution and reacted at pH of 8. PEGDA was dissolved in urea-PBS to a final concentration of 280 mg/mL and then slowly added to the fibrinogen solution and allowed to react for 3 h in the dark at room temperature. After the reaction, PEGylated fibrinogen was extracted with acetone, then centrifuged to remove acetone, and finally dissolved in urea-PBS again for dialysis. The product was dialyzed in sterile PBS over 24 h in the dark at 4°C and then stored at −80°C. Protein content was determined to be 12.5 mg/mL using a BCA protein assay kit (Thermo Fisher). PEGylation yield was calculated to be 98.1%.
Cell encapsulation in PEG-fibrinogen microspheres
Cell encapsulation in PF hydrogel microspheres was achieved through a custom-developed microfluidic polydimethylsiloxane (PDMS) system. The PDMS mold was created with the Sylgard 184 silicone elastomer kit (Dow Corning) by pouring the mixture of base and cure components into a polystyrene dish containing the preassembled microfluidic channel mold. The mixture was subsequently degassed and heat cured at 60°C for 2 h. After curing, the microfluidic channel mold was disassembled. The microfluidic PDMS mold was sonicated in 70% ethanol before and after each use.
Before cell encapsulation, PF hydrogel precursor solution was prepared by addition of 1% (v/v) of 10% (w/v) pluronic F68 (Sigma) in PBS, 1% (v/v) of 10 mM of eosinY photoinitiator (Fisher Scientific) in PBS, 1.5% (v/v) triethanolamine (Acros Organics), and 0.39% (v/v) of N-vinylpyrrolidone (Sigma). Pluronic F68 is an amphiphilic block copolymer that is commonly used as a surfactant to stabilize the emulsion of microencapsulation, which narrows the size distribution of the microspheres. It has been previously shown that the addition of a low concentration of pluronic had a negligible effect on the hydrogel degradation profile. 31 ECFCs were detached by trypsinization as already described, centrifuged, and resuspended in hydrogel precursor solution to a cell density of 10 million cells per mL.
Cell encapsulation and hydrogel photocrosslinking (Fig. 1) were conducted in a biosafety cabinet. The microfluidic device had two inlets and one outlet; the PF hydrogel precursor with suspended cells flowed from the top inlet, and mineral oil flowed from the bottom inlet using syringe pumps. When the two streams met at the junction, microspheres were formed due to emulsification, and the cell-encapsulated microspheres were crosslinked by a 2.7W full-spectrum visible light (Prior Lumen 200). A mirror was placed behind the microfluidic device near the outlet to aid in crosslinking by reflecting the light that passed through the device. The microspheres were washed down from the outlet with pre-warmed Dulbecco's modified Eagle's medium (DMEM) by using a third syringe pump setting at 22 mL/h. The microspheres were then washed twice with DMEM by centrifugation at 200 g for 3 min to remove the residual mineral oil and cultured in endothelial growth medium on a collagen-coated well plate at 37°C with 5% CO2.
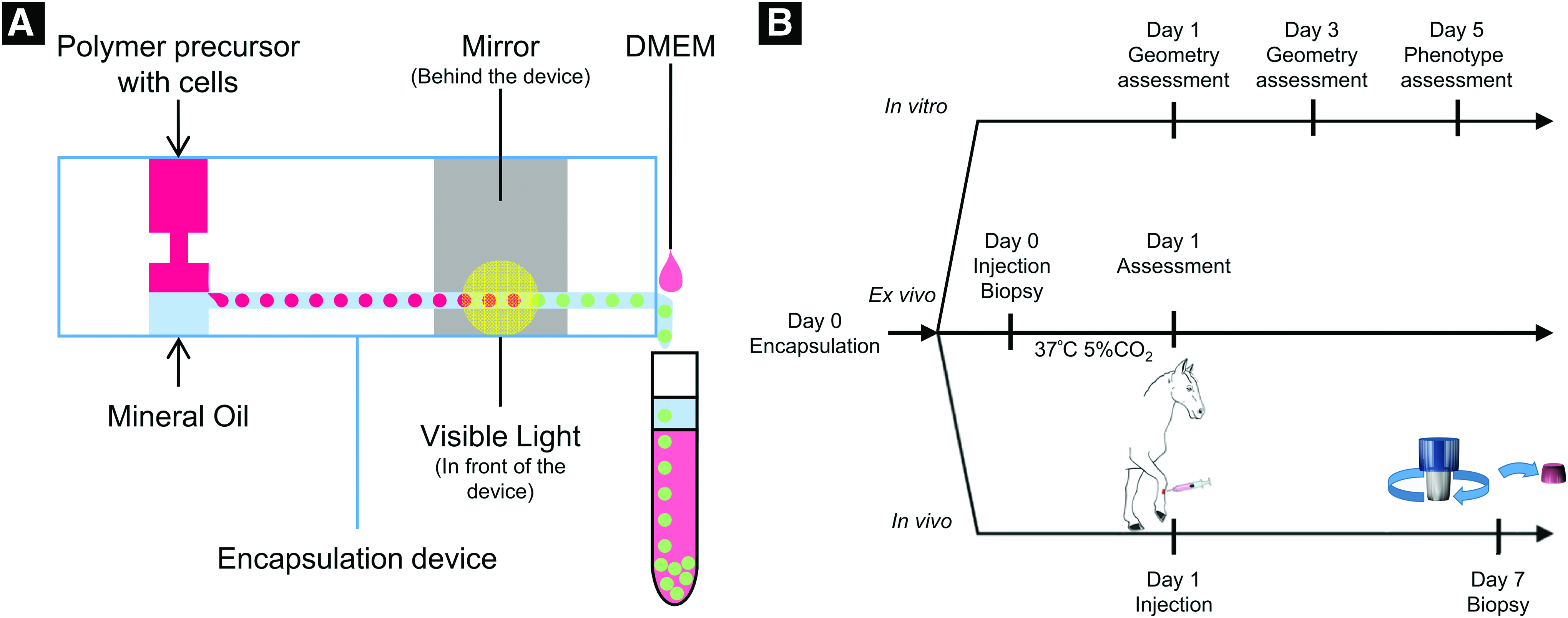
Post-encapsulation cell viability assay
Cell viability after encapsulation was assessed using Live/Dead viability/cytotoxicity (Invitrogen) kits for Live/Dead staining. Microspheres collected immediately after encapsulation were incubated for 30 min with calcein AM and ethidium homodimer-1, and then Z-stack-images were acquired with fluorescence microscopy. Three regions with same size (250 × 250 μm) were randomly selected on each microsphere using ImageJ and viability was then assessed by counting Live/Dead-stained cells through the optical slices along the z-axis for ∼550 μm.
Microsphere geometry characterization
The uniformity of the microspheres was evaluated by measuring their maximum diameter and roundness 1 and 3 days after cell encapsulation. Three batches of microspheres with at least 30 microspheres per batch were measured, and the measurements were performed using ImageJ.
Microsphere stiffness
To measure the Young's modulus of the hydrogel microspheres, they were subjected to compression testing under physiological conditions using the MicroSquisher (CellScale). Briefly, ECFCs encapsulated in microspheres were cultured for 1 or 3 days before mechanical testing. These microspheres were then loaded onto the MicroSquisher platform, maintained at 37°C in PBS, preconditioned for compression testing, and made to undergo cycles of compression and relaxation at a rate of 2.5 μm/s with a minimum of 15% strain. The force–displacement data obtained from the stress were converted to stress–strain curves, and the lower portion of the curve (5–15% strain) was used to estimate the Young's moduli of microspheres.
Outgrowth cell phenotypic characterization
To assess the phenotype of the encapsulated cells, microspheres were cultured on collagen-coated well plates, and cells growing out from microspheres onto the well plate were harvested and subcultured once to obtain sufficient cells for characterization. To ensure the ECFC phenotype was not impacted by the number of subcultures, ECFCs that had the same passage number in culture but did not undergo encapsulation, were used for comparison.
Uptake of 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate-labeled acetylated low-density lipoprotein
The ability of ECFCs to take up 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate-labeled acetylated low-density lipoprotein (DiI-Ac-LDL) was evaluated. Ten micrograms per milliliter of DiI-Ac-LDL (Biomedical Technologies) in pre-warmed medium was added to the ECFCs followed by incubation for 6 h at 37°C and 5% CO2. After incubation, uptake of DiI-Ac-LDL by ECFCs was imaged using fluorescent microscopy.
In vitro tubule formation assay
ECFCs were seeded onto a 96-well plate (30,000 cells/well) containing Matrigel (Corning, 50 μL/well), which had been incubated for 15 min before cell seeding. Tubule formation was assessed after 5 h of incubation at 37°C and 5% CO2 by light microscopy.
Immunofluorescence analysis
Equine ECFCs pre- and post-encapsulation were evaluated for the expression of cell markers von Willebrand factor (vWF), CD14, and CD105 with indirect immunofluorescence assay (IFA) as previously published. 32 ECFCs were fixed with 4% paraformaldehyde (PFA) solution and rinsed with PBS solution. For analysis of expression of the intracellular protein vWF, ECFCs were permeabilized with PBS-T containing 0.2% TritonX 100 (Sigma) in PBS for 30 min, and then blocked with 3% horse serum at 4°C overnight. The fixed cells were then incubated at room temperature for 1 h with primary antibodies diluted in 3% horse serum as follows: rabbit anti-human vWF (Dako) at 1:100, mouse anti-horse CD14 at 1:200 (Wagner Laboratory, Cornell University 33 ), and mouse anti-human CD105 at 1:200 (AbD Serotec).
After incubation, cells were washed with PBS-T before the application of secondary antibodies. Secondary antibodies were diluted in 3% horse serum and incubated with cells at room temperature in the dark for 1 h as follows: Alexa Fluor 488-conjugated goat anti-rabbit immunoglobulin G (IgG) at 1:400 for vWF and Alexa Fluor 488-conjugated goat anti-mouse IgG at 1:400 for CD14 and CD105. Cells were counterstained with 4′,6-diamidino-2-phenylindole (DAPI), washed with PBS, mounted on glass slides with ProLong Gold antifade reagent (Life Technologies), and imaged with fluorescent microscopy.
Flow cytometry
To assess the influence of the encapsulation process on ECFC phenotype, flow cytometry was used to quantify expression of vWF, CD14, CD105, and uptake of DiI-Ac-LDL. For DiI-Ac-LDL uptake, cells were incubated with medium containing 10 μg/mL of DiI-Ac-LDL at 37°C for 6 h. After incubation, cells were rinsed with medium and PBS, detached, centrifuged, and fixed in suspension with 4% PFA for 20 min at room temperature, followed by a rinse with PBS. The cells were then stored in 3% horse serum in PBS.
For vWF staining, detached cells were fixed, permeabilized, and blocked with 10% horse serum at 4°C overnight. Then, cells were incubated with primary antibodies at room temperature for 1 h, rinsed with blocking solution, and incubated in secondary antibodies at room temperature for 1 h in the dark. For CD14 and CD105 staining, detached cells were incubated with primary antibodies at 4°C for 1 h, rinsed with blocking solution, incubated in secondary antibodies at room temperature for 1 h in the dark, rinsed with blocking solution, and then fixed. The same concentrations of the primary and secondary antibodies were used as for IFA. Cells incubated with the secondary antibody only were used to set the gating for negative population. Cells were resuspended in 3% horse serum in PBS and filtered using Flowmi tip strainers (Bel-Art) before flow analysis. A total of 10,000 events were collected for each sample, and a BD Accuri C6 flow cytometer (BD Biosciences) was used to analyze forward scatter versus side scatter plots. Flow cytometry gates were set to select for live cells and to eliminate doubled cells, dead cells, and debris.
Cell viability after injection through a syringe and needle
To further assess the potential of the microspheres as vehicles for injectable cell delivery, the effect of shear stress during injection on cell viability was studied. Microspheres were suspended in cold equine serum, with a viscosity of 1.909 cP at 14°C, loaded into 1 mL luer lock syringes, and sheared through 18-, 20-, and 23-gauge needles, respectively, at 1 mL/min. 34 The XTT Cell Viability Assay Kit (Biotium) was then used to evaluate cell viability and proliferation. Following the injection simulation, microspheres containing ECFCs were aliquoted into a 96-well plate with one microsphere per well. One hundred microliters of prewarmed medium and 25 μL of XTT working solution were then added to each well. After incubation for 18 h at 37°C, absorbance signal of the sample was measured with a microplate reader (BioTek). Four separate trials were performed; viability was assessed for five microspheres per condition per trial and data for each trial normalized with respect to control optical density.
Microspheres for subcutaneous injection
Ex vivo cell delivery and survival of encapsulated ECFCs
A cadaver limb from an adult horse collected immediately after euthanasia (for reasons unrelated to this study) was used to evaluate cell delivery ex vivo. The hair of the dorsomedial aspect of the metacarpus was clipped and two 6.25 cm2, full-thickness wounds were created using a surgical template. Trypan blue-stained PEGDA microspheres in saline or ECFCs encapsulated into PF microspheres in serum were injected (600 μL) subcutaneously at the wound edge. Injections were made through an 18-gauge × 1″ needle on a 1 mL luer lock syringe. Trypan blue-stained PEGDA microspheres were directly visualized. A full-thickness section of skin and subcutaneous tissue surrounding the area of injection were removed using a scalpel blade and placed in cell culture media and incubated overnight at 37°C. Subcutaneous tissue was bluntly dissected at 24 h and visualized with fluorescent and phase-contrast microscopy.
In vivo injection and cell tracking of autologous ECFCs encapsulated in PF microspheres
Three adult horses were used to evaluate delivery of encapsulated, autologous ECFCs labeled with Qtracker 655 (Invitrogen) to full-thickness wounds created on the distal limb. Autologous ECFCs were labeled with 4 μM Qtracker 655 according to the manufacturer's instructions before encapsulation. The equine distal limb wound model was created in a similar manner to previous studies.35–37 Horses were kept free in individual box stalls for the duration of the study and allowed ad libitum access to grass hay and water. For the surgical procedure, analgesia and sedation were provided with detomidine hydrochloride (0.01 mg/kg; IV) and butorphanol tartrate (0.04 mg/kg; IV), and local anesthesia was performed using 2% mepivacaine hydrochloride. The surgical sites were clipped and aseptically prepared, and two 6.25 cm2, full-thickness wounds were created on the dorsal aspect of each metacarpus and metatarsus using a sterile wound template and a No. 15 scalpel blade.
Twenty-four hours after surgery, the edges of two wounds per horse were injected subcutaneously using 18-gauge 1″ needles on 1 mL luer lock syringes with autologous ECFCs encapsulated in PF microspheres diluted into equine serum (600 μL). Each injection of cell-laden microspheres contained ∼2 × 106 ECFCs in PF microspheres. All four edges of the wound were injected for a total of 8 × 106 ECFCs in PF microspheres per wound. As the wound model was also being utilized for a larger, ongoing treatment trial with ECFCs, the remaining wounds were treated in duplicate with autologous ECFCs (2 × 106), PF microspheres, and equine serum only and evaluated weekly until complete healing (∼6 weeks). One week after injection, the horses were sedated, local anesthesia performed, and then the lateral leading edge of each was biopsied using a 6 mm punch biopsy instrument. One biopsy sample was placed in optimal cutting temperature compound and snap frozen in liquid N2 cooled isopentane and the other sample was formalin fixed and paraffin embedded for immunohistochemical analysis as part of the wound healing study. Frozen tissues were cryosectioned (20 μm), placed on glass slides, fixed in 4% PFA, stained with DAPI, and imaged with confocal microscopy.
Statistical analysis
Data are represented as mean ± standard deviation. All statistical analyses were performed using Minitab 17 Statistical Software (Minitab, Inc.). After verifying equal variances using the F-test, the Student's t-test was used for comparisons between two groups. After checking for normality of distribution, one-way analysis of variance followed by the Tukey–Kramer honest significant difference (HSD) test for multiple comparisons was used to evaluate statistical significance between multiple groups. Unless otherwise indicated, p < 0.05 was considered statistically significant.
Results
High cell viability and uniform microsphere geometry post-encapsulation
Microspheres formed using the custom-built PDMS molded microfluidic device were highly uniform and provided a suitable microenvironment for cell proliferation and survival. Following microsphere encapsulation, ECFC viability was very high (96.8 ± 1.4%) as shown in Figure 2A–D; a Z-stack of a representative Live/Dead-stained microsphere is shown in Supplementary Movie S1 (Supplementary Data are available online at

High microsphere uniformity and cell viability post-encapsulation.
Increased elastic modulus and outgrowth from microspheres indicating cell proliferation post-encapsulation in PEG-fibrinogen microspheres
The stiffness of the microspheres was assessed in terms of elastic modulus. The elastic modulus of the microspheres with encapsulated ECFCs increased significantly from 141 ± 10 Pa on day 1 to 354 ± 62 Pa on day 3 (Fig. 2I). This increase in modulus correlated with a visual increase in total cell number and reorganization of the ECFCs within the microspheres (Figs. 2F and 3).
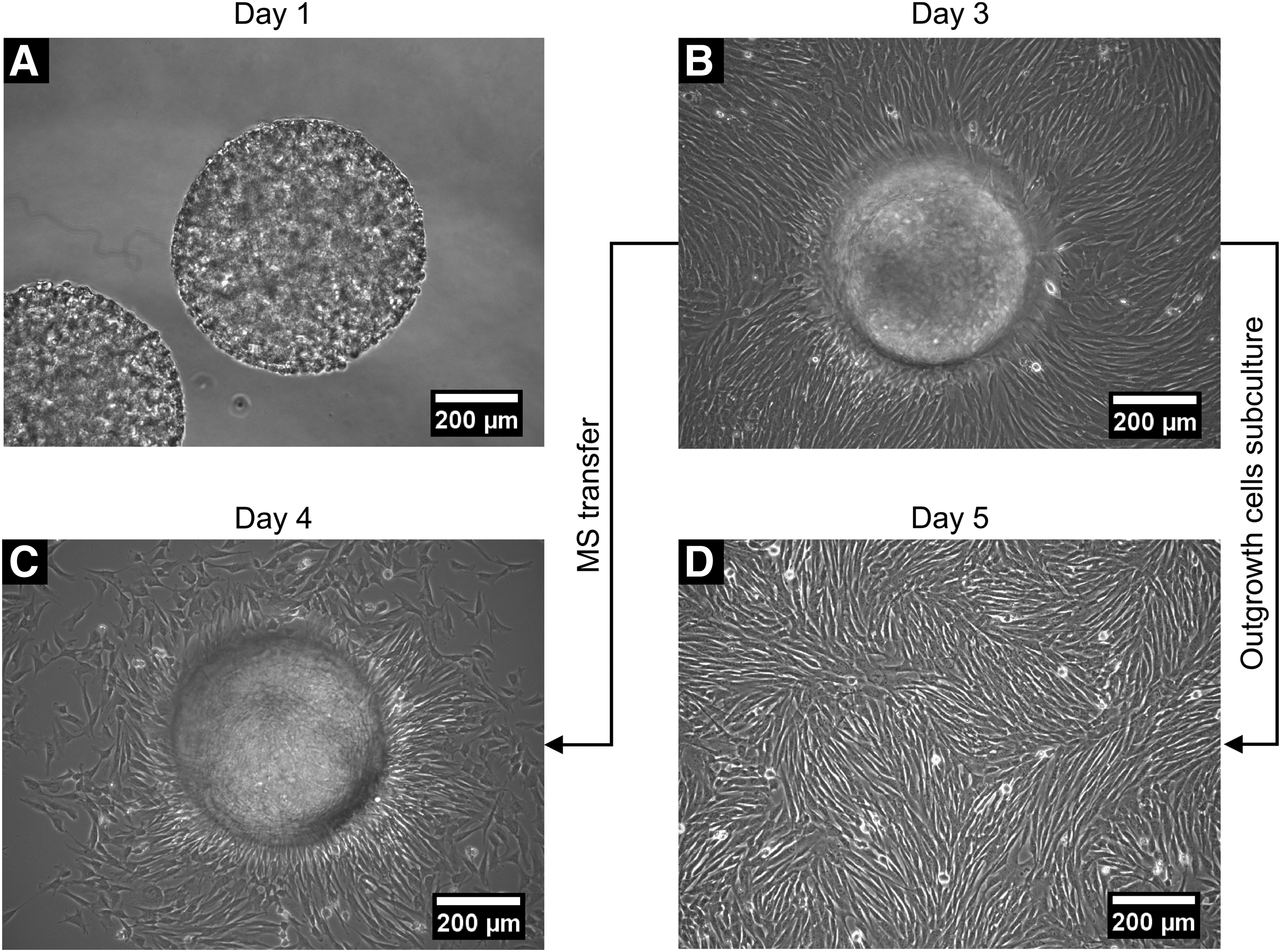
Phase-contrast images showing ECFC migration, and proliferation phenotypes were maintained post-encapsulation.
In addition to maintaining a high level of viability, the ECFCs also remained highly proliferative post-encapsulation. On day 1, the encapsulated ECFCs were observed to align and cover the surface of the microspheres (Fig. 3A). As ECFCs continued to remodel the microspheres, a confluent monolayer of ECFCs was observed on the bottom of the well plate by day 3 indicating active cell migration and proliferation (Fig. 3B). After the ECFCs and microspheres were trypsinized and transferred to a new well plate, ECFCs were again observed on the bottom of the well plate around the replated microspheres after just one day (Fig. 3C). In addition, the confluent monolayer of outgrowth ECFCs observed on day 3 was passaged, and the passaged outgrowth ECFCs again formed a confluent monolayer at an equivalent rate to encapsulated control ECFCs, indicating that the ECFCs maintained their highly proliferative nature following microsphere encapsulation (Fig. 3D).
Outgrowth ECFCs maintain their phenotype
ECFCs have the same phenotypic characteristics pre- and post-encapsulation. Both outgrowth ECFCs and nonencapsulated ECFCs (only cultured on TCPS flasks) were able to form tubular networks on Matrigel, take up DiI-Ac-LDL, and express vWF as shown in Figure 4A–H. Endothelial function and expression of markers previously used to characterize equine ECFCs 32 were evaluated quantitatively in both groups of ECFCs using flow cytometry. For ECFCs that were only cultured on TCPS flasks (nonencapsulated), 98.6 ± 1.4% of cells demonstrated uptake of DiI-Ac-LDL, 99.8 ± 0.2% of cells expressed vWF, 99.8 ± 0.2% expressed CD105, and 98.1 ± 1.5% expressed CD14 (Fig. 4I). In outgrowth ECFCs from the microspheres, 99.0 ± 1.6% of cells demonstrated uptake of DiI-Ac-LDL, 99.3 ± 0.6% of cells expressed vWF, 98.5 ± 1.7% expressed CD105, and 98.4 ± 1.0% expressed CD14 (Fig. 4I). No significant differences were found between the two groups.
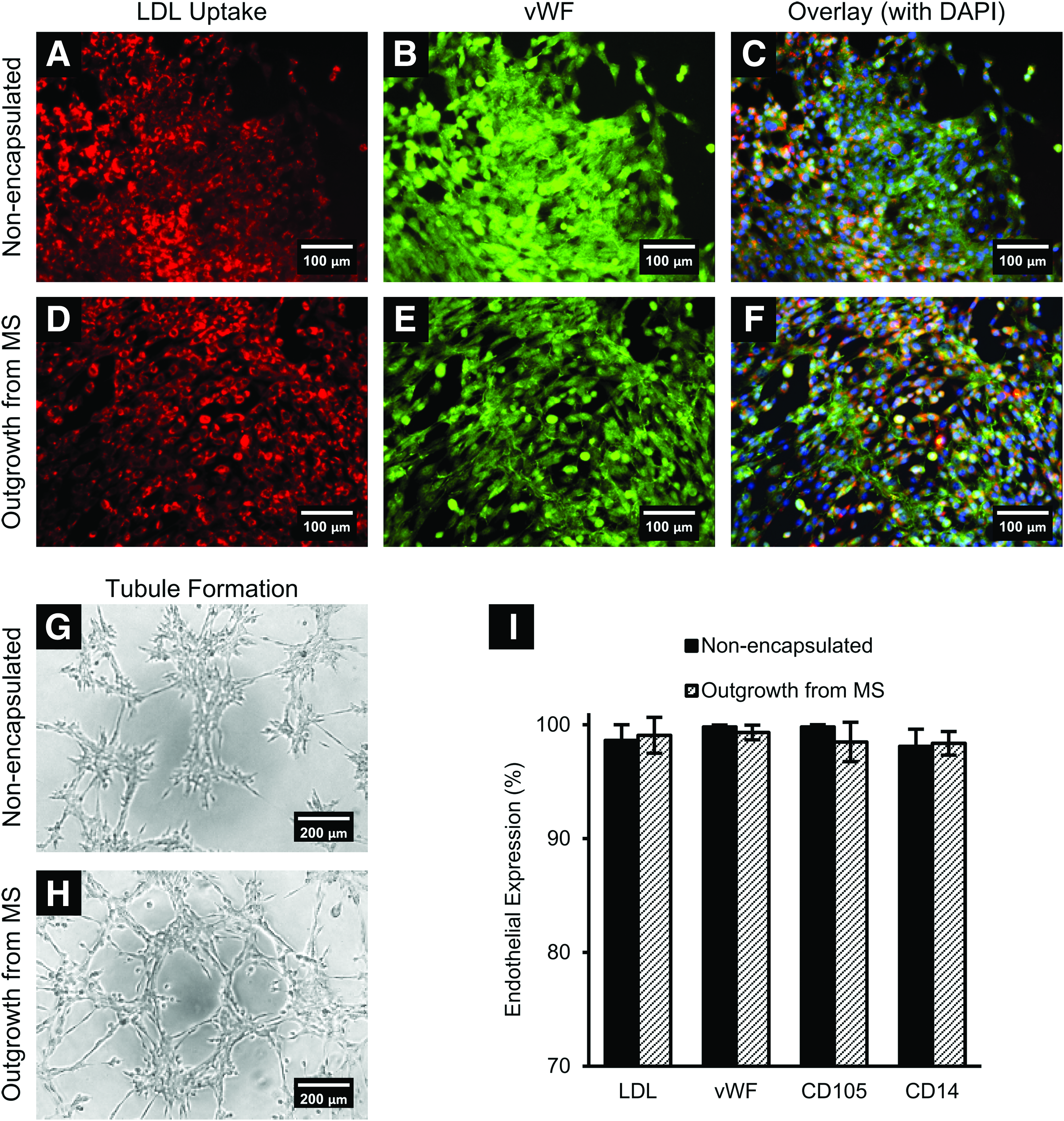
ECFCs maintained their endothelial phenotype after encapsulation and culture in PF microspheres. Outgrowth ECFCs from microspheres showed similar endothelial phenotype compared to nonencapsulated ECFCs in terms of DiI-Ac-LDL uptake, vWF expression
Microspheres are retained and encapsulated cells are viable in tissue after subcutaneous injection and shear through different gauge needles
To evaluate the potential of using hydrogel microspheres for cell injection therapy, the retention of microspheres in tissue and the viability of encapsulated ECFCs after shear through different needle gauges were examined ex vivo. There was no statistical difference in viability of encapsulated ECFCs as quantified by XTT assay after shear through 18-, 20-, and 23-gauge needles (Fig. 5A). Cell-free microspheres that were created with PEGDA and stained with trypan blue were injected subcutaneously into the edge of a wound created on an equine cadaver limb using 18-gauge 1″ needles. The injected microspheres remained in the subcutaneous tissue at the wound edge without any visually obvious breakdown or deformation of the microspheres (Fig. 5B).
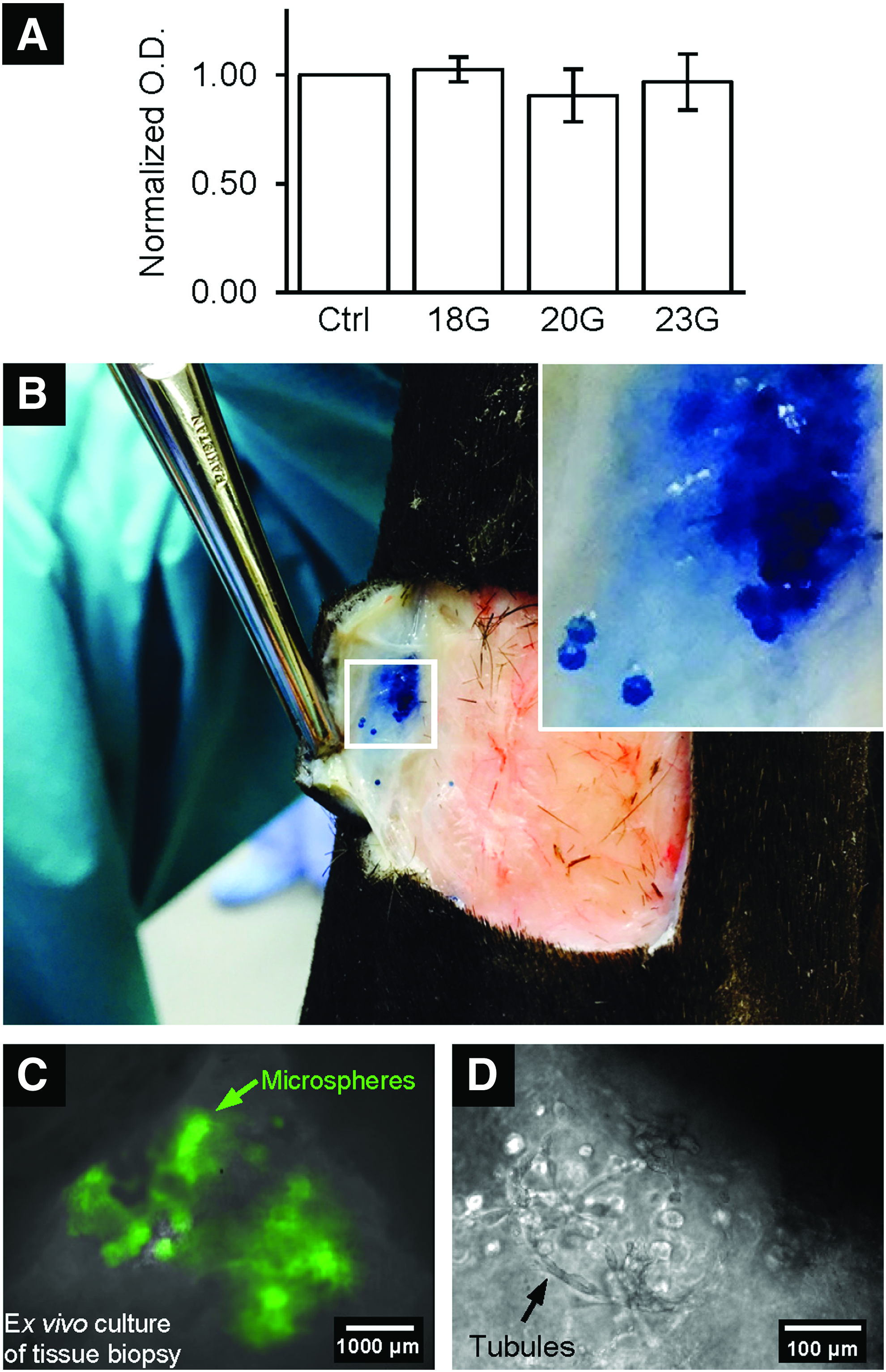
Injected microspheres supported cell delivery.
An ex vivo experiment was then performed to evaluate viability and migration from the microspheres of the encapsulated cells when injected into and surrounded by host tissue. After one day of ex vivo culture at 37°C, subcutaneously injected microspheres remained present as a group within the tissue as indicated by the green autofluorescence of eosin Y (Fig. 5C). Furthermore, encapsulated ECFCs became elongated and formed tubules along the surface of the microspheres (Fig. 5D). This observation was similar compared to the in vitro study shown in Figure 3.
PEG–fibrinogen-encapsulated ECFCs are retained at the injection site and beginning to migrate out of PF 7 days after in vivo subcutaneous injection
An in vivo study was performed in three horses to evaluate cell retention and outgrowth of the encapsulated ECFCs to the surrounding host tissue. Autologous ECFCs were labeled with Qtracker 655 (red, Fig. 6A) and then encapsulated at a concentration of 10 million cells per mL of PF precursor solution, which was necessary to achieve the desired dose of 2 million cells for each injection. For each horse, ECFCs were encapsulated for four injections per wound with two wounds per animal plus an in vitro control, meaning that 24 million cells were encapsulated. After microfluidic system setup, encapsulation took ∼12 min per 2 million cell injection.

Qtracker-labeled ECFCs remained visible for cell tracking post-encapsulation and 1 week after in vivo injection.
Following rinsing and resuspension, cell-laden microspheres were injected into the edges of distal limb wounds in three adult horses. One week postinjection of the microspheres into the edge of a wound, the Qtracker-labeled ECFCs (red) were identified in the biopsy of all three horses as shown in a representative biopsy sample in Figure 6B. The injected microspheres were still present in one horse as shown by the green autofluorescence. There was evidence of migration of some ECFCs from the microspheres to the surrounding host tissue suggesting viable cells as indicated by the white arrows. The PF-encapsulated ECFCs were verified to be retained at the injection site and demonstrated outgrowth 1 week after subcutaneous injection.
Discussion
For veterinary and human clinical applications, delivery of therapeutic cells by injection has the advantage of being minimally invasive. In this study, we demonstrate the rapid encapsulation of equine ECFCs in PF microspheres using a custom-built microfluidic encapsulation system; verify the uniformity of resulting microspheres; confirm the maintenance of cell viability, proliferative capacity, and phenotype; and establish the ability of these microspheres to be used for delivery of cells by injection for autologous cell therapy.
Having used the equine wound healing model, we have tested the ability of our cell encapsulation system to be used for the large numbers of cells (2 million cells per injection, 8 million cells per wound) that are likely necessary to achieve therapeutic benefit. The achieved cell concentration of 10 million cells per mL of hydrogel precursor solution is substantially higher than has been reported for other microencapsulation systems, which was necessary to maintain a reasonable injection volume (200 μL of microspheres suspended in serum for total of 600 μL). Also, the time required for encapsulation is short (12 minutes for 2 million cells), which was necessary to produce sufficient numbers of encapsulated cells within a reasonable time frame for this large animal model and eventual clinical use.
Utilization of hydrogel technologies has received much attention for regeneration of vasculature and ultimately healing of a range of vascular diseases and disorders. 38 Previous work has primarily focused on formation of macroscopic tissues and polymerization of the cell/hydrogel precursor in situ, both of which would frequently require a surgical approach for many clinical applications.39,40 As an alternative approach, cell microencapsulation for the purpose of therapeutic cell delivery has been investigated as recently reviewed.41–43 While technically challenging to produce, hydrogel microspheres have many advantages for use in cell delivery compared to cell/scaffold implantation methods. Microspheres are able to be more widely distributed across a wound or tissue than cell-supporting scaffolds with other geometries, enabling maintenance of short diffusion distances from the host tissue while simultaneously providing structural support to enhance cell survival and retention following implantation. Furthermore, microspheres can be injected at one or more sites, increasing their flexibility for clinical use and reducing surgical trauma from implantation. 44 Microfluidic systems have better control in producing microspheres with a narrow size distribution compared to traditional methods, including bulk emulsification, electrostatic dripping, extrusion methods, and hydrodynamic dripping. 16 By utilizing an emulsification technique in microfluidic systems, hydrogel microspheres can be fabricated with various stream geometries, including flow-focusing, T-junction, terrace-like, and coflowing. 45 However, typical cell encapsulation in microspheres using microfluidic devices has been limited by low cell density, which makes delivery of sufficient cell numbers in reasonable volumes for therapeutic applications challenging. 16 The microfluidic device presented here is capable of encapsulating ECFCs at a high cell density of 10 million cells per mL, which is essential for delivering a reasonable volume of microspheres while having an effective number of cells for cell therapy. In addition, a unique molding approach was used to fabricate these devices. Microfluidic devices are generally fabricated using photolithography, which is time-consuming and requires expensive microfabrication facilities. When using photolithography, the channel height of the microfluidic devices is restricted due to limitations in the thickness of photoresist that can be coated onto the silicon wafer. This study employs a novel method for microfluidic device fabrication that does not rely on the use of photolithography, thereby enabling devices to be readily produced with larger channel geometries and without the need for expensive microfabrication facilities. These devices, therefore, can be used for encapsulating cells in hydrogel microspheres that are larger in size than those typically produced using microfluidic chips. In addition, the use of emulsification and the microfluidic system together achieved highly reproducible and tightly controllable microsphere size and geometry.
We used an equine wound model to study in vivo cell delivery for this study. There are only a few studies using biomaterial scaffolds to optimize the function of stem cells in equine regenerative medicine, most of which have involved musculoskeletal disease.46–48 This is the first study to use PF hydrogels for cell delivery/tissue healing/regenerative studies in veterinary medicine. These horses were part of a larger, ongoing study on wound healing, the first step of which was to confirm that cells were delivered and retained at the site of interest as reported in this study. Full-thickness skin wounds of the distal limb that heal by second intention are common injuries in horses, and delayed healing and formation of exuberant granulation tissue (EGT) are frequent complications. At the time of the wound, cutaneous blood flow is disrupted, leading to altered tissue perfusion and reduced oxygen delivery.
The decrease in blood flow is more pronounced and longer in duration in equine distal limb wounds than in body wall wounds. 37 The extent and duration of loss of blood supply after the initial wound may determine the amount of hypoxia and tissue inflammation that leads to formation of EGT rather than normal healing. Although once EGT forms it is highly vascular, not all of the blood vessels that form are equally functional, and many occluded microvessels have been identified in leg wounds with EGT. 35 Ideally, a healing wound regains vascular supply as soon as possible and the vessels that form need to be functional so that local hypoxia and subsequent inflammation do not occur. The potential benefit of ECFC treatment in other models of poor wound healing due to poor vascularization49,50 prompted the use of autologous equine ECFCs for use in the current study.
Promoting rapid vascularization is one of the biggest challenges in using engineered tissues for enhancing wound healing and treating disease. 51 While ECFCs, or late EPCs, have been described in the literature as the only subtype of EPCs responsible in building vessels, evidence has also shown that early EPCs promote angiogenesis through a paracrine mechanism.4,52 Recent studies have also suggested that adult stem cell therapy provides the majority of benefits through paracrine effects rather than direct tissue replacement.53,54 Although assessing the use of ECFCs alone to achieve rapid wound healing is beyond the scope of this study, the presented method of combining ECFCs with engineered biomaterials allows the direct delivery and retention of ECFCs, as well as the appropriate growth factors and signaling molecules needed, in the area of interest.
Encapsulation of ECFCs in PF hydrogels has great potential in tissue engineering and clinical applications. PF was developed to create a controllable, degradable, and biofunctional 3D scaffold for cell culture. 26 While PEG provides high biocompatibility and versatile physical structure, fibrinogen provides biological functionality, including protease degradation capability 55 and cell-adhesion motifs. 56 This is to the best of our knowledge the first study encapsulating autologous ECFCs in PF and demonstrating the support of ECFC survival, maintenance of phenotype and outgrowth for use in injectable cell therapy for tissue regeneration applications. Encapsulated ECFCs were observed to cover the surface of the microspheres by day 3 showing that they are able to migrate through and remodel the PF hydrogel microsphere. Whereas microsphere size remained the same through day 3, the modulus increased and microspheres supported cell growth.
One of the most common challenges in cell therapy is the inability to benefit from all the injected therapeutic cells. Direct injection of cells alone is commonly used in human and veterinary regenerative medicine and may minimize the complexity of the delivery; however, the cells are not retained or do not migrate to the area of interest. Studies have shown that intracoronary delivery of cardiomyocytes by saline injection caused cells to appear in the lung, liver, and spleen. 11 The loss of cells to other organs through the vascular system may also impose other complications. For instance, intravenous delivery of ECFCs may contribute to tumorigenesis, atherosclerosis, or retinopathy, so this strategy should be followed clinically with utmost caution. 15 In addition, ischemic areas may have low blood supply, so intravenous delivery of ECFCs may not reach the site of injury. Therefore, local delivery of cells that results in high retention is critical. By injecting the microspheres with labeled cells directly at the site of injury, we were able to verify the presence of cells 1 week after injection.
Conclusions
We established the ability to encapsulate ECFCs in PF hydrogel microspheres using a unique microfluidic device. High concentrations of cells could be rapidly encapsulated making the system practical for use in cell encapsulation for large animal therapeutic cell delivery. The resulting cell-laden microspheres were highly uniform in size and geometry with strong consistency between batches. The ECFCs had high viability and continued to proliferate post-encapsulation. Outgrowth ECFCs from microspheres exhibited the same phenotype as ECFCs maintained in traditional monolayer culture. As a proof of concept, microspheres encapsulated with Qtracker-labeled ECFCs were injected into an equine distal limb wound model, and cell retention at the injection site and migration of outgrowth ECFCs into the host tissue was observed 1 week after injection confirming that this is a feasible system for injectable cell delivery.
Footnotes
Acknowledgments
Supported, in part, by the Grayson Jockey Club Research Foundation, a joint AU-CMB/NSF EPSCoR (NSF-EPS-1158862) summer research fellowship (W.J.S.), the American Heart Association [AM HEART-14SDG18610002 (W.J.S., Y.T., and E.A.L.)], and the National Science Foundation Chemical, Bioengineering, Environmental, and Transport Systems program [NSF-CBET-1150854 (E.A.L.)]. The authors thank Ms. Ashley Sharpe for assistance and Mr. Brennen Reece for providing the horse illustration.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.