
Abstract
Cardiac tissue engineering by means of synthetic or natural scaffolds combined with stem/progenitor cells is emerging as the response to the unsatisfactory outcome of approaches based solely on the injection of cells. Parenchymal and supporting cells are surrounded, in vivo, by a specialized and tissue-specific microenvironment, consisting mainly of extracellular matrix (ECM) and soluble factors incorporated in the ECM. Since the naturally occurring ECM is the ideal platform for ensuring cell engraftment, survival, proliferation, and differentiation, the acellular native ECM appears by far the most promising and appealing substrate among all biomaterials tested so far. To obtain intact scaffold of human native cardiac ECM while preserving its composition, we compared the decellularized ECM (d-ECM) produced through five different protocols of decellularization (named Pr1, Pr2, Pr3, Pr4, and Pr5) in terms of efficiency of decellularization, composition, and three-dimensional architecture of d-ECM scaffolds and of their suitability for cell repopulation. The decellularization procedures proved substantially different. Specifically, only three, of the five protocols tested, proved effective in producing thoroughly acellular d-ECM. In addition, the d-ECM delivered differed in architecture and composition and, more importantly, in its ability to support engraftment, survival, and differentiation of cardiac primitive cells in vitro.
Introduction
A
The ECM is by definition an ideal biological scaffold material. As it is custom designed and manufactured by the resident cells of each tissue and organ, it provides the architecture for multicellularity10,11 and delivers both mechanical and chemical signals to cells. Furthermore, the ECM is maintained in dynamic equilibrium with the cellular components of the organ, responding to physiological and pathological conditions through qualitative and quantitative changes in its composition.
Over the last decade, scientists in the field of regenerative medicine have dedicated significant effort to developing biological scaffolds obtained through decellularization of ECM, and decellularized cardiac ECM is emerging as the most promising material to serve as a template to support stem cell-based cardiac regeneration.
Decellularized ECM (d-ECM) from myocardium is obtained through physical or chemical methods or a combination of both. 12 Several chemical reagents and physical methods have been used thus far, although the optimal combination is yet to be found. The ideal method of decellularization should ensure the complete removal of cells and cell remnants while preserving ECM composition and its three-dimensional architecture. Unfortunately, partial loss of ECM components is inevitable with both chemical and physical methods. Therefore, the best procedure for decellularization of an organ is one that removes all cellular components while preserving as much of the ECM as possible.
With the final aim of constructing patches of human myocardium for cardiac repair, we selected three protocols for the decellularization of human myocardium or pericardium and used them to prepare acellular scaffolds of human myocardium. In addition, we decellularized human myocardium by adapting a popular protocol used for the decellularization of the whole heart and also used a fifth novel protocol that has not been previously tested. Finally, we compared the five d-ECM scaffolds obtained to evaluate the effectiveness of decellularization, ECM organization, and ECM composition. Moreover, we isolated human cardiac primitive cells (hCPCs) to evaluate d-ECM scaffold cytocompatibility and its ability to support hCPC differentiation in vitro.
Materials and Methods
Cardiac specimens
Explanted hearts of patients (n = 10, mean age 49.5 ± 4,7) undergoing heart transplantation because of end-stage heart failure associated with ischemic cardiomyopathy were obtained from Cardiothoracic Surgery at the Monaldi Hospital (Naples, Italy). Samples were harvested from macroscopically uninjured areas of the free wall of the left ventricle, and snap-frozen or enzymatically digested to isolate cardiac primitive cells (hCPCs). All patients provided written, informed consent for use of heart tissue for experimental studies and specimens were collected, without patient identifiers, following protocols approved by Monaldi Hospital and in conformity with the principles outlined in the Declaration of Helsinki.
Myocardium sectioning
Frozen specimens were mounted on a cryostat chuck on the endocardial surface using tissue freezing medium (Leica Microsystems, Wetzlar, Germany). Then, 350-μm-thick sections were cut by a Leica CM1950 cryostat (Leica Microsystems), discarding the sections with the epicardium to obtain cryosections including only the myocardium. Cryosections were collected in sterile cell culture dishes and stored at −80°C until use for either decellularization or as controls for molecular analysis. Three cryosections for each specimen were fixed in formalin for paraffin embedding.
Decellularization
Four previously published decellularization protocols, referred to as Pr1, Pr2, Pr3, and Pr4, were tested. Pr1, 13 Pr2, 14 and Pr3 15 were protocols specifically established for the decellularization of human cardiac thin slices, while Pr4 was adapted from a popular protocol used for whole heart perfusion decellularization. 16 In addition, several changes were made to Pr4 to shorten the procedure. We also developed and tested a fifth protocol, named Pr5.
Pr1
According to the “three-step protocol,” 350-μm-thick sections were incubated in lysis buffer (10 mM Tris, 0.1% wt/vol ethylenediaminetetraaceticacid [EDTA], pH 7.4) for 2 h, followed by solubilization in sodium dodecyl sulfate (SDS, 0.5% wt/vol in phosphate-buffered saline [PBS]) for 6 h. After washing with PBS (three times for 10 min plus overnight), residual DNA was removed by incubation in fetal bovine serum (FBS) for 3 h at 37°C. Following a second washing step, samples were stored in PBS plus 0.5% penicillin/streptomycin (all chemicals were purchased from Sigma-Aldrich, St. Louis, MO) at 4°C for up to 2 weeks before recellularization. All steps were performed on an orbital shaker at room temperature.
Pr2
As described by Godier-Furnémont, myocardial sections were lysed for 2 h in 10 mM Tris buffer and 0.1% wt/vol EDTA and then solubilized for 6 h in 0.5% SDS with orbital mixing. Successively, sections were washed in PBS, incubated in PBS containing 50 U/mL DNase (Sigma-Aldrich) and 1 U/mL RNase (Sigma-Aldrich), and washed in Hanks' Balanced Salt Solution (HBSS) with orbital mixing to remove detergent and cell debris.
Pr3
In agreement with Mirsadraee, but substituting 350-μm-thick myocardial sections for pericardial samples, specimens were washed for 90 min in PBS containing protease inhibitors (aprotinin, 10 KIU/mL; 0.1% w/v EDTA), changing the wash buffer every 30 min, and then treated with hypotonic buffer (10 mM Tris-HCl; pH 8.0) for 16 h at 4°C in the presence of protease inhibitors. Next, cryosections were incubated for 24 h in 0.1% (w/v) SDS in hypotonic buffer at room temperature and then washed in sterile PBS for three periods of 30 min. Tissues were then incubated in reaction buffer containing 50 U/mL DNase I and 1 U/mL RNase A in 10 mM Tris-HCl (all from Sigma-Aldrich) at pH 7.5 for 3 h at 37°C. Finally, samples were washed in PBS for three periods of 30 min. All steps were performed on an orbital shaker.
Pr4
Following the protocol used by Ott for decellularization of the whole heart, myocardial sections were incubated in 1% SDS (Sigma-Aldrich) in deionized water for 12 h. This was followed by 15 min of washing in deionized water and 30 min of incubation with 1% Triton X-100 (Sigma-Aldrich) in deionized water. Sections were then rinsed for 124 h in antibiotic solution containing 100 U/mL penicillin and 50 U/mL streptomycin (Sigma-Aldrich) and amphotericin B (Sigma-Aldrich) in PBS. All steps were performed with agitation on an orbital shaker.
Pr5
The fifth protocol has not been previously tested and was developed by modifying Pr4. Specifically, myocardial sections were incubated in 1% SDS and 1% Triton X-100 (both from Sigma-Aldrich) in deionized water for 24 h. The sections were then rinsed for 24 h in antibiotic solution containing 100 U/mL penicillin and 50 U/mL streptomycin (Sigma-Aldrich) and 0.25 μg/mL amphotericin B (Sigma-Aldrich) in PBS and then for an additional 30 min in sterile bidistilled water. All steps were performed with agitation on an orbital shaker.
After decellularization, three cryosections for each specimen were stored in Ham's F12 medium (Sigma-Aldrich) at 4°C, three decellularized cryosections were fixed in formalin for paraffin embedding and histological analysis, and all other cryosections were stored at −80°C until use for molecular analysis.
Histochemistry and immunohistochemistry
Cryosections, native and decellularized, were routinely fixed in 10% neutral-buffered formalin, dehydrated in a graded series of alcohol, cleared in xylene, infiltrated, and embedded in paraffin according to standard protocols. Paraffin blocks were then sliced into serial 5-μm-thick sections and placed on poly-lysine-coated glass slides for histological or histochemical staining and evaluation.
Sections were deparaffinized, rehydrated, and stained for Masson's trichrome and Sirius Red, as previously described.17,18 In addition, sections were stained with hematoxylin and eosin, Periodic Acid-Schiff (PAS) Morel-Maronger modified, and Gomori's paraldehyde-fuchsin using specific staining kits (all from Bio-Optica, Milan, Italy) and following the manufacturer's protocol. Hematoxylin and eosin and Masson's trichrome stainings were used to evaluate the effectiveness of decellularization procedures and the architecture of the decellularized myocardium, Sirius Red was used to detect collagen, and PAS and Gomori stain kits were used to detect glycoproteins and elastic fibers, respectively.
Immunodetection of fibronectin, tenascin, and laminin on tissue sections was performed using primary antibodies against human fibronectin, tenascin, and laminin (all from Sigma-Aldrich) and the indirect immunoperoxidase technique by using the UltraVision LP Detection System HRP Polymer and DAB Plus Chromogen (Thermo Scientific, Waltham, MA), according to the manufacturer's protocol, to reveal the presence and localization of antigen–antibody complexes.
The structure of native and decellularized tissues and the efficacy of the decellularization procedure were evaluated and documented by at least three independent observers using a light microscope DM2000 Led (Leica Microsystems) equipped with an ICC50HD camera (Leica Microsystems).
Quantitative measurement of DNA content
Genomic DNA was extracted from frozen native and decellularized cardiac cryosections, using the AllPrep DNA/RNA Mini Kit (Qiagen, Hilden, Germany), according to the manufacturer's instructions.
Briefly, tissue samples were lysed and homogenized in a highly denaturing buffer containing guanidine isothiocyanate. The buffer immediately inactivated DNases and RNases, thus ensuring isolation of intact DNA and RNA. Lysates were then passed through an AllPrep DNA spin column, which, in combination with the high-salt buffer, allowed selective and efficient binding of genomic DNA. The columns were washed and pure, ready-to-use, and DNA was then eluted.
DNA concentration was determined by measuring the absorbance at 260 nm using a NanoDrop1000 (Thermo Scientific), and the gDNA band was visualized with agarose electrophoresis. Data were averaged and expressed as the mean value ± standard deviation (SD) of nanogram of DNA per milligram of tissue for each protocol.
Quantitative measurement of sulfated glycosaminoglycan and collagen
Native and decellularized cryosections were dissolved in a digestion buffer containing papain or acid, and the obtained tissue extracts were used in the Blyscan or Sircol Insoluble quantitative dye-binding assay (Biocolor Ltd., Carrickfergus, United Kingdom) for the analysis of sulfated glycosaminoglycans (sGAGs) or insoluble collagen content, respectively. Assays were performed according to provided assay manuals. In the Sircol assay, 20 mg powdered ECM for each sample was digested with the fragmentation reagent supplied by the manufacturer at 65°C for 2 h. The digested ECM samples were then assayed following the instructions supplied by the manufacturer, and each assay was performed in triplicate. The absorbance was read at 600 nm using the BioPhotometer (Eppendorf, Hamburg, Germany) and analyzed with KaleidaGraph software (Synergy Software, Reading, PA). In the Blyscan assay, 20 mg of ECM for each sample was digested using the papain extraction reagent supplied by the manufacturer at 65°C for 3 h. The digested ECM samples were then assayed following the instructions supplied by the manufacturer, and each assay was performed in triplicate. The absorbance was read at 600 nm using the BioPhotometer (Eppendorf) and analyzed with KaleidaGraph software (Synergy Software). Data were averaged and expressed as the mean value ± SD of μg of sGAG or collagen per milligram of wet tissue for each protocol.
Growth factor array
Three sections of d-ECM for each protocol were lysed in lysis buffer containing Tris-HCl, 5 Mm EDTA, Triton X-100, 0.01 M dithiothreitol, phenylmethylsulfonyl fluoride, and protease inhibitor cocktail. Protein concentration was quantified by a Bradford assay, and then 60 μg of each tissue lysate was assayed in the Human Growth Factor Array C1 (RayBiotech, Norcross, GA) to simultaneously detect 41 targets. The procedure was performed in strict accordance with the manufacturer's directions. Briefly, five array membranes were blocked with blocking buffer for 30 min at room temperature, and then, 1 mL of each sample was added to a membrane and incubated at room temperature for 2.5 h. Next, the membranes were washed three times in wash buffer I, two times in wash buffer II, and then incubated with biotin-conjugated antibody overnight at 4°C. After further washes, membranes were incubated for 2 h at room temperature with horseradish peroxidase-conjugated streptavidin and washed a final time to remove unbound reagents. All incubation steps were performed with agitation on an orbital shaker. Membranes were then developed with the detection buffer, exposed to film, and processed by autoradiography. As suggested by the manufacturer, visual comparison of the array images was sufficient to determine differences in protein presence and relative protein expression. In addition, numerical comparison of the signal densities of growth factors known to influence or modulate stem cell fate was performed strictly according to the guidelines for data extraction supplied with the array protocol. Briefly, spot signal densities from the scanned images of arrays were obtained using ImageJ densitometry software (
Cell isolation and culture
Cardiac tissue samples harvested from the free wall of the left ventricles of explanted human hearts were dissected, minced, and enzymatically disaggregated by incubation in 0.25% trypsin for 6 h at 4°C and in 0.1% (w/v) collagenase II (both from Sigma-Aldrich) for 30 min at 37°C. The digestion was stopped by adding double volume of HBSS supplemented with 10% FBS. This preparation was further disaggregated by pipetting, and tissue debris and cardiomyocytes were removed by sequential centrifugation at 100 g for 2 min, passaged through a 40-μm cell strainer (BD Biosciences, Franklin Lakes, NJ), and centrifugation at 400 g for 5 min.
The obtained cell population was depleted of fibroblasts by immunomagnetic cell sorting based on the Miltenyi Biotec protocol. Briefly, the cell suspension was previously incubated with antifibroblast MicroBeads (Miltenyi Biotec) to magnetically label fibroblasts. Cells were then loaded onto the MACS column (Miltenyi Biotec) and placed in the magnetic field of MACS separator (Miltenyi Biotec) to retain labeled fibroblasts within the column and allow unlabeled cells running through. Hence, the negative fraction of hCPCs was collected and used for repopulating d-ECM cryosections.
In vitro assay of d-ECM biocompatibility
d-ECM cryosections were stored at 4°C in Ham's F12 medium (Sigma-Aldrich) supplemented with 10% FBS (Sigma-Aldrich), 5% horse serum (Sigma-Aldrich), 10 ng/mL basic fibroblast growth factor (bFGF; Peprotech, Rocky Hill, NJ), and 50 IU/mL penicillin G/streptomycin (Sigma-Aldrich) cell culture medium until they were repopulated with hCPCs. Before cell seeding, cryosections were mounted on sterile 35-mm cell culture dishes, sterilized by exposure to ultraviolet radiation for two cycles of 20 min each, and rehydrated for 5 days in an incubator at 37°C with 5% CO2, with the same cell culture medium used for storage. After rehydration, 4.5 × 105 hCPCs were seeded onto d-ECM sections and cultured under standard culture conditions in the same medium. As a control, 7.5 × 104 cells were seeded on 35-mm uncoated cell culture dishes.
Cells were checked daily for viability and morphology by microscopic observation with an Olympus CKX41 (Olympus Corporation, Tokyo, Japan) inverted microscope equipped with a ColorView IIIu digital camera (Olympus Corporation), and, as control cells reached 100% confluence, all cells were kept in culture for three more days to allow differentiation. To successively evaluate any difference in differentiation potential in the presence of d-ECM from different protocols, cells were collected after trypsinization and processed for real-time polymerase chain reaction (PCR) analysis.
To determine the viability of hCPCs and cytotoxicity of the d-ECM obtained from the five decellularization protocols in vitro, cryosections were mounted on sterile 96-well plates, sterilized by exposure to ultraviolet radiation for two cycles of 20 min each, and rehydrated for 5 days in an incubator at 37°C with 5% CO2 with the same cell culture medium used for storage. After rehydration, 2 × 104 hCPCs were seeded onto d-ECM sections and cultured under standard culture conditions in the same medium. 2 × 104 hCPCs were seeded onto uncoated wells to serve as control for intrinsic cell death, as apoptosis. Cell death and survival rates were assessed using trypan blue stain (Lonza, Walkersville, MD) as previously described. 19 Twenty-four and forty-eight hours after seeding, medium was collected and centrifuged at 400 g for 5 min to obtain the fraction of the detached dead cells that stained blue with trypan, as dead cells do not exclude the dye. Dead cells were then counted using a hemocytometer and expressed as percentage of total seeded cells. Seventy-two hours from seeding, cells were collected by trypsinization, and cell viability was assessed by counting alive and dead cells using a hemocytometer with standard trypan blue (0.4% in PBS) exclusion staining. The numbers of viable and dead cells were expressed as the mean percentage (%) of the total number of cells ± SD.
Gene expression analysis by real-time quantitative polymerase chain reaction
Gene expression analysis was performed as previously described. 20 Briefly, total RNA was extracted from hCPCs cultured on decellularized myocardial sections using Isol-RNA Lysis Reagent (5Prime, Hamburg, Germany) according to the manufacturer's instructions. RNA was dissolved in RNase-free water, and the final concentration was determined using a NanoDrop 1000 spectrophotometer (Thermo Scientific). All RNA samples were quality checked and were considered suitable for gene expression profiling experiments. Then, 100 ng of RNA from each sample was reverse transcribed into cDNA with a QuantiTect Reverse Transcription Kit (Qiagen), following the protocol provided by the supplier. Gene expression was quantified by real-time quantitative polymerase chain reaction (qPCR) using 5Prime Hot MasterMix (5Prime). The primer assays used in this study are included in Table 2. DNA amplification was carried out using Mastercycler ep realplex4S (Eppendorf), and detection was performed by measuring the binding of the fluorescence dye SYBR Green I to double-stranded DNA (dsDNA). All samples were tested in triplicate with the housekeeping gene (GAPDH) to correct for variations in RNA quality and quantity. Melt curve analyses were conducted to assess uniformity of product formation, primer/dimer formation, and amplification of nonspecific products. Linearity and efficiency of PCR amplification were assessed using standard curves generated by increasing amounts of cDNA. Comparative quantification of target gene expression in the samples was performed based on the cycle threshold (Ct) normalized to the housekeeping gene and to Pr5, which served as a calibrating sample, using the ΔΔCt method.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 5.0 (GraphPad Software, La Jolla, CA). Data obtained for the collagen, sGAG, growth factor array, trypan blue exclusion test, and qPCR measurements were analyzed using one-way analysis of variance with Bonferroni post-tests. All data are expressed as the mean ± SD. A value of p < 0.05 was used to identify any statistically significant differences.
Results
Evaluation of the decellularization procedure effectiveness
Snap-frozen cardiac specimens harvested from the free wall of the left ventricle of human hearts were cryosectioned, and 350-μm-thick sections were decellularized according to the five different procedures (Table 1). Hematoxylin and eosin and Masson's staining were used, along with quantitative measurements of DNA content to evaluate the effectiveness of each decellularization protocol. Cell remnants were still visible in specimens decellularized with Pr3 (Fig. 1), indicating ineffective decellularization. Accordingly, analysis of DNA content confirmed the presence of residual dsDNA (96.7 ± 12.23 ng/mg wet tissue) above the currently accepted standard of <50 ng dsDNA per milligram of decellularized biomaterials 12 in the d-ECM obtained according to Pr3. In d-ECM obtained with the other four protocols, the amounts of dsDNA met the established criterion. However, while Pr2, Pr4, and Pr5 yielded d-ECM scaffolds containing dsDNA amounts well below the limits (9.0 ± 2.1, 17.33 ± 2.22, and 4.7 ± 0.71 ng/mg wet tissue, respectively), the d-ECM from Pr1 contained 46.33 ± 5.3, which sometimes exceeded the limit considering the variability (Fig. 2). Moreover, d-ECM obtained with Pr5 contained the lowest amount of residual dsDNA, and differences obtained with the Bonferroni post hoc test between the mentioned protocol and all other protocols were significant (p < 0.05).
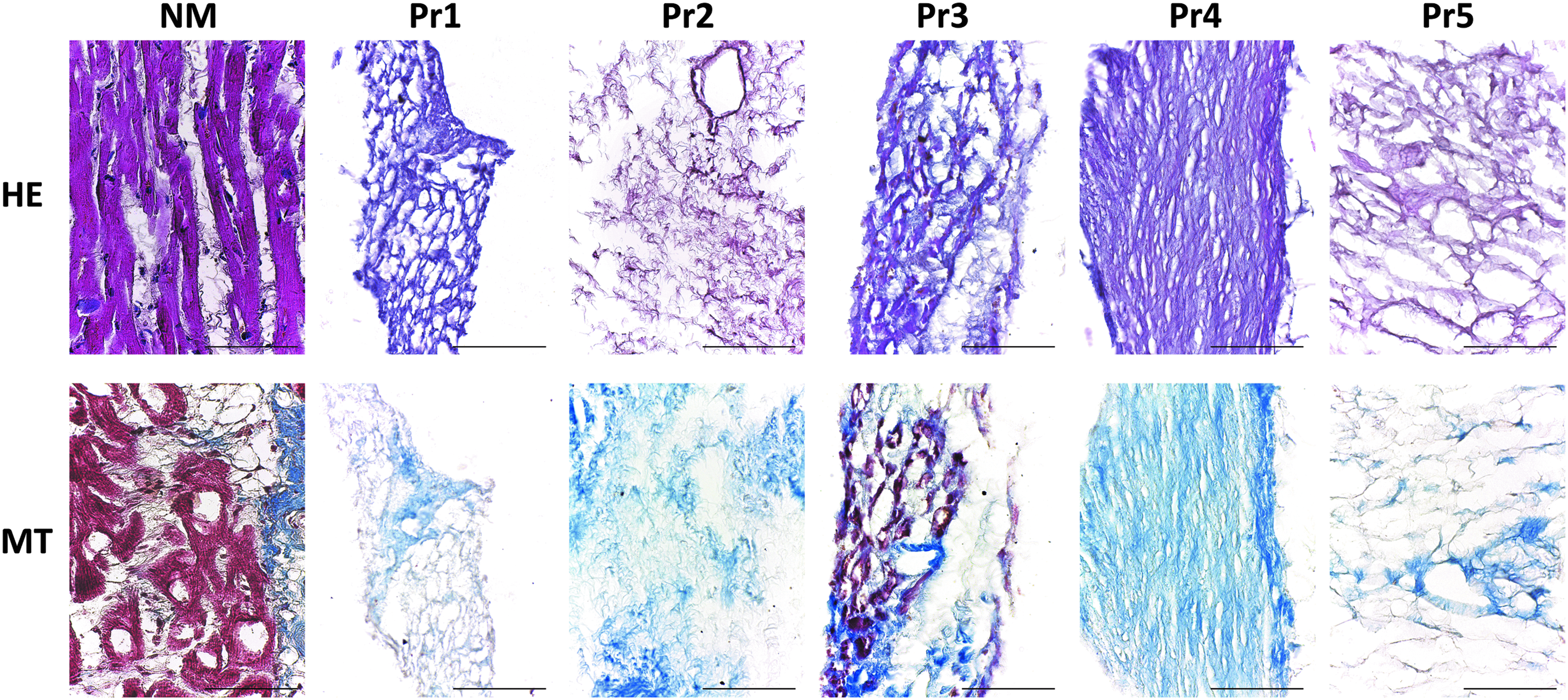
Representative images of HE staining and MT staining on 350-μm-thick cryosections of NM or of myocardium decellularized according to different decellularization protocols (from Pr1 to Pr5). HE stains the nuclei dark violet and the cytoplasm pink or purple, MT stains the myocytes red and the collagen blue. Scale bar length is 50 μm. HE, hematoxylin and eosin; MT, Masson's trichrome; NM, native myocardium; Pr1, protocol 1; Pr2, protocol 2; Pr3, protocol 3; Pr4, protocol 4; Pr5, protocol 5.

Measurement (left) and quantification (right) of DNA content in cryosections of NM or of myocardium decellularized with different procedures (from Pr1 to Pr5). Each value expresses the mean of 10 samples (n = 10) ± SD. Asterisks are indicators of the p-value as follows: significant (*p ≤ 0.05 vs. Pr5) and extremely significant (***p ≤ 0.001 vs. Pr5). SD, standard deviation.
RT, room temperature; EDTA, ethylenediaminetetraaceticacid; SDS, sodium dodecyl sulfate; PBS, phosphate-buffered saline; FBS, fetal bovine serum; HBSS, Hanks’ Balanced Salt Solution.
Analysis of d-ECM composition
Specific histological staining methods and immunohistochemistry were used to evaluate tissue architecture, differentiate ECM components, and assess their retention after decellularization. Sirius Red, PAS, and Gomori's stainings documented the retention of collagen, noncollagenous proteins, and elastic fibers, respectively, in d-ECM from all protocols, although more preserved architectures of d-ECM mesh surrounding regular empty spaces occupied by cardiomyocytes before decellularization occurred were evident in d-ECM obtained with Pr4 and Pr5 (Fig. 3). Immunodetection of fibronectin, tenascin, and laminin confirmed that ECM composition and distribution were preserved to a large extent. In particular, this was more noticeable in d-ECM from Pr4 and Pr5, which showed better preserved architecture (Fig. 4).

Representative images of Sirius Red, Periodic Acid-Schiff (PAS) Morel-Maronger modified, and Gomori's paraldehyde-fuchsin histochemical stainings on 350-μm-thick cryosections of NM or of myocardium decellularized according to different decellularization protocols (from Pr1 to Pr5). Sirius Red stains the collagen fibers red, PAS stains the glycoproteins violet, and Gomori's stains the elastic fibers pink. Scale bar length is 50 μm.

Representative images of immunodetection of noncollagenous ECM proteins in 350-μm-thick cryosections of NM or of myocardium decellularized with different protocols (from Pr1 to Pr5). The brown color shows the presence and the distribution of fibronectin, tenascin, or laminin. Scale bar length is 50 μm. ECM, extracellular matrix.
Blyscan and Sircol Insoluble assays allowed quantitative measurements of sGAG and collagen content in d-ECM and demonstrated the retention of both components in scaffolds obtained with all five protocols (Fig. 5). Accordingly, d-ECM obtained through Pr3 retained higher amounts of collagen (16.080 ± 0.161 μg/mg wet tissue), followed by d-ECM obtained with Pr2 (10.736 ± 0.40 μg/mg wet tissue), Pr5 (10.622 ± 0.041 μg/mg wet tissue), and Pr4 (9.741 ± 0.040 μg/mg wet tissue), while d-ECM obtained with Pr1 retained the lowest amount of collagen (4.909 ± 0.039 μg/mg wet tissue). Differences between protocols as calculated with Bonferroni's multiple comparison test were statistically significant (p < 0.05), with the only exception for the comparison between Pr2 and Pr5 (Fig. 5A).
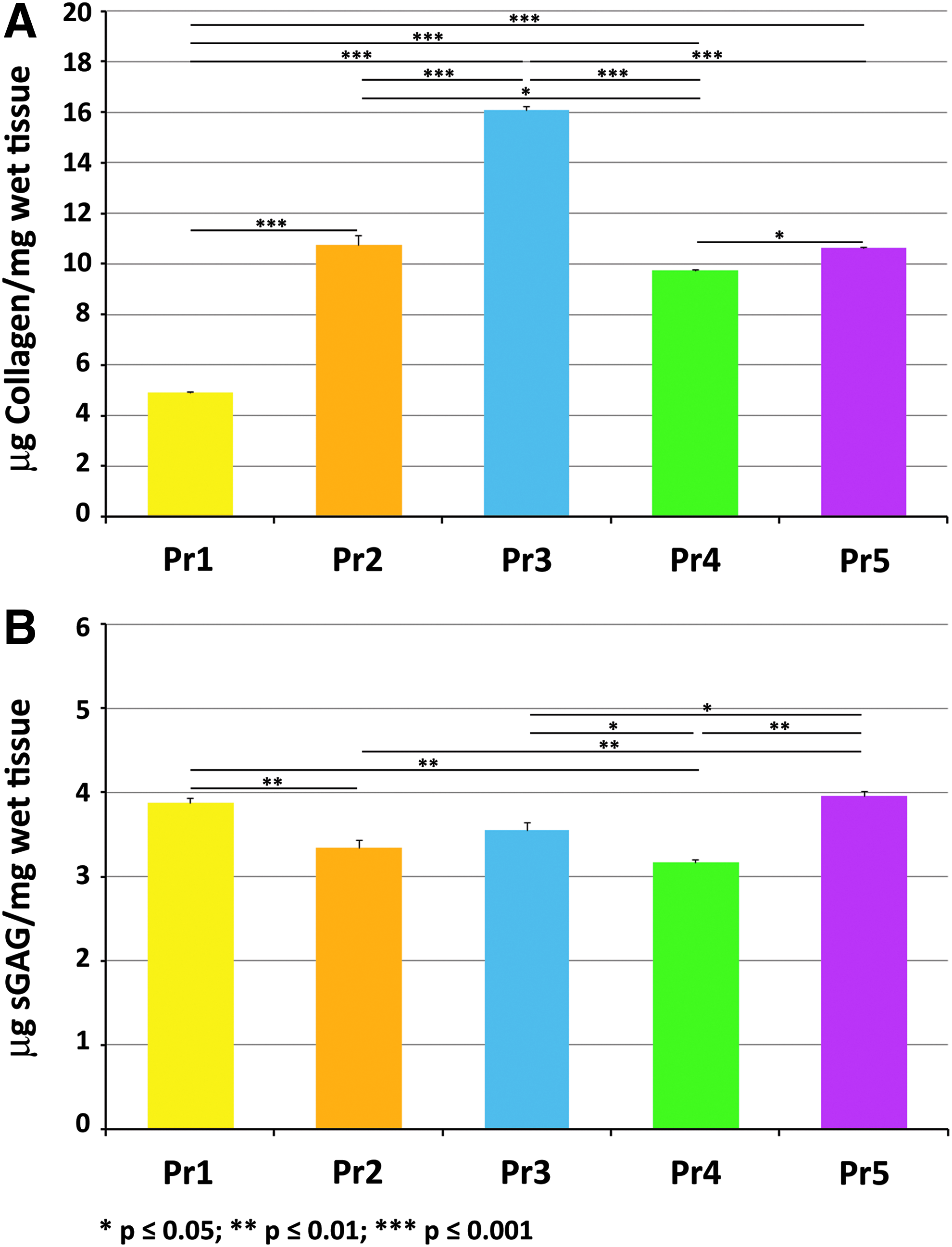
Quantification of collagen
Regarding sGAG, Blyscan assay revealed that d-ECM obtained with all protocols retained sGAG. In particular, the highest content of sGAG was retained by d-ECM obtained with Pr5 (3.96 ± 0.06 μg/mg wet tissue), followed by those decellularized with Pr1 (3.875 ± 0.06 μg/mg wet tissue), Pr3 (3.556 ± 0.09 μg/mg wet tissue), Pr2 (3.344 ± 0.09 μg/mg wet tissue), and Pr4 (3.173 ± 0.03 μg/mg wet tissue). Statistically significant differences (p < 0.05) were observed between d-ECM obtained from Pr5 and d-ECM from Pr2, Pr3, or Pr4 (Fig. 5B).
Because GAGs protect growth factors against proteolysis,21,22 we investigated the retention of growth factors by d-ECM by protein array that simultaneously detects the presence of 41 targets between growth factors and their receptors. Analysis revealed that growth factors and receptors of growth factors were still stored in d-ECM decellularized with Pr1, Pr2, Pr3, and Pr4, while d-ECM scaffolds obtained with Pr5 contained growth factors but lost growth factor receptors. Specifically, growth factors such as FGF-4, FGF-6, insulin-like growth factor (IGF)-2, and stem cell factor (SCF) were not affected by the decellularization procedure for any protocol, while different protocols had different effects on the retention of bFGF, epidermal growth factor (EGF), hepatocyte growth factor (HGF), macrophage-colony stimulating factor (M-CSF), platelet-derived growth factor (PDGF)-AA, PDGF-AB, and PDGF-BB, transforming growth factor (TGF), vascular endothelial growth factor (VEGF), and their receptors (Fig. 6). Notably, d-ECM obtained from Pr5 still contained bFGF, EGF, PDGF, but lost HGF, TGF, VEGF, and growth factor receptors, while d-ECM obtained from Pr1, Pr2, Pr3, and Pr4 retained EGF, VEGF, and receptors for EGF, PDGF, SCF, and VEGF. In addition, the d-ECM obtained from Pr1 and Pr3 retained significantly (p < 0.05) higher amounts of FGF-7 and significantly (p < 0.05) higher amounts of EGF, M-CSF, and related receptors, while the d-ECM obtained from Pr4 retained significantly (p < 0.05) higher amounts of PDGF-AA and VEGF and related receptors (Supplementary Figs. S1 and S2; Supplementary Data are available online at
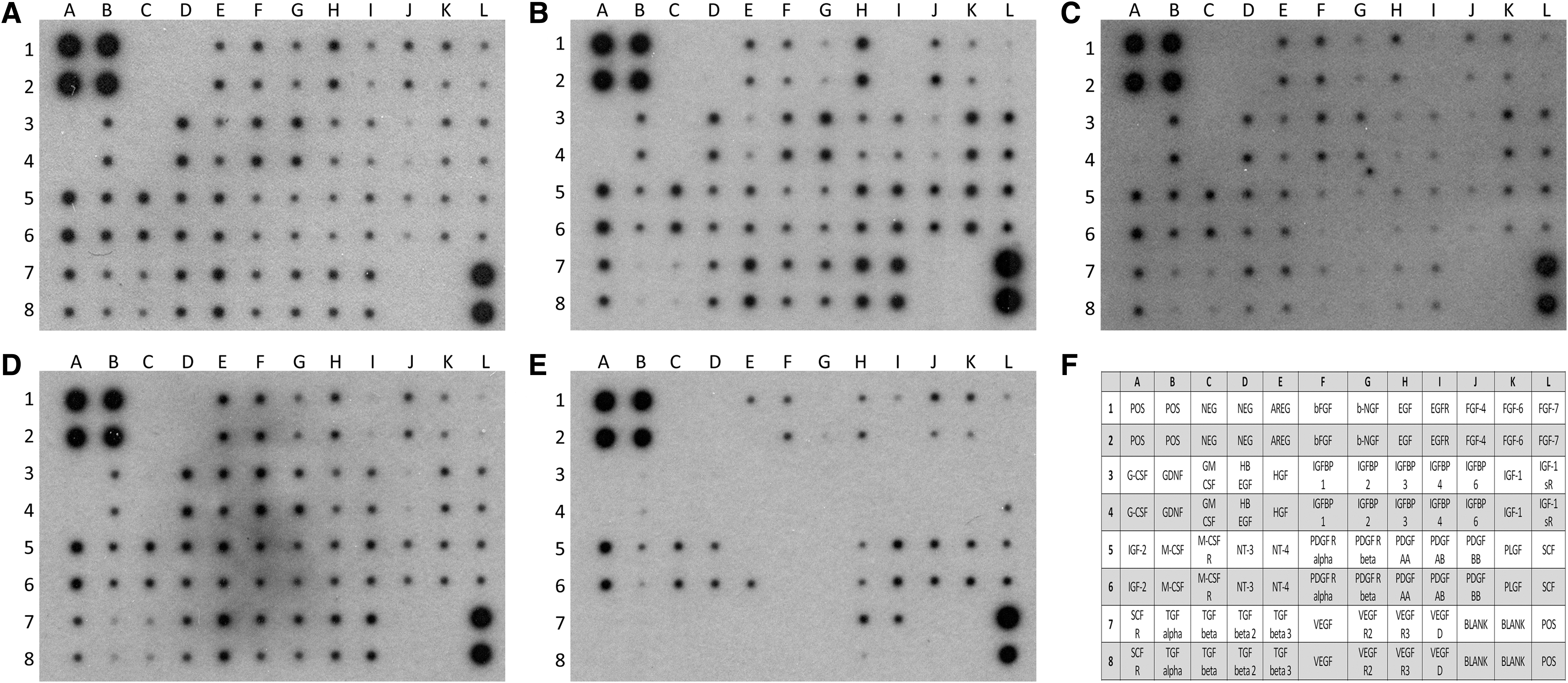
Profile of growth factors and receptors for growth factors retained by decellularized ECM after decellularization procedure performed with the five different protocols.
In vitro assessment of d-ECM biocompatibility
The d-ECM must be biocompatible to be effectively used for regenerative purposes. Thus, to assess cytocompatibility of d-ECM in vitro, we seeded and cultured hCPCs on 350-μm-thick sections of d-ECM obtained with all five protocols. Although we planned to culture cells for at least 3 days for cell viability assay and for at least 7 days for gene expression analysis, cultures on d-ECMs obtained from Pr1 and Pr3 were discarded after 48 h, as most of the cells had died, detached, and were floating in the culture medium. In contrast, hCPCs were successfully cultured for up to 7 days on d-ECM obtained with Pr2, Pr4, and Pr5.
Cell viability was quantified using a trypan blue assay. The dead cells with damaged plasma membrane were stained with trypan blue due to the uptake of this dye and the inability to exclude it. Twenty-four hours after seeding, a considerable number of hCPCs seeded on the d-ECM from Pr1 and Pr3 had died and detached from the d-ECM (48.12 ± 14.71 and 53.26 ± 15.43, respectively) and a larger fraction had died and detached after 48 h, reaching almost the total number of the seeded cells (89.11 ± 6.53 and 93.38 ± 3.77, respectively) (Supplementary Fig. S3A). In contrast, the d-ECM from Pr2, Pr4, and Pr5 better supported cell viability as death rate after 24, 48, and 72 h stayed steadily low on d-ECM from Pr5 (0.52 ± 0.11, 0.48 ± 0.30, 0,73 ± 0.49, respectively) and reached the highest rate on the d-ECM from Pr2 (2.85 ± 1.25, 10.45 ± 1.56, 3.49 ± 1.28, respectively). The d-ECM from Pr4 showed an acceptable survivability, although after 48 and 72 h the death rate of hCPCs seeded on the d-ECM from Pr4 was significantly (p < 0.05) higher than the death rate on the d-ECM from Pr5 (6.11 ± 1.24 after 48 h, 2.50 ± 0.79 after 72 h) (Supplementary Fig. S3B). After 72 h, all cells were trypsinized and live cells were counted along with dead cells. Supplementary Figure S3 shows the mean viability percentage of cells seeded on d-ECMs from Pr2, Pr4, and Pr5. Again, the d-ECM from Pr5 showed the best survivability as the mean percentage of live cells resulted significantly (p < 0.01) higher (42.74 ± 15.91, 87.47 ± 7.11, 97.43 ± 1.02 on the d-ECM from Pr2, Pr4, and Pr5, respectively) (Supplementary Fig. S3C).
To evaluate the ability of the d-ECM to serve as a viable environment to support the differentiation of hCPCs, we analyzed the expression of genes involved in the differentiation of cardiomyocytes (MEF2C, ACTC1), endothelial cells (ETS1, FVIII), and smooth muscle cells (GATA6, ACTA2) by qPCR (Table 2). Due to the cytotoxicity of d-ECM decellularized in accordance with Pr1 and Pr3, we compared gene expression of hCPCs cultured on d-ECM from Pr2, Pr4, and Pr5. Gene expression analysis revealed that d-ECMs obtained from different decellularization procedures supported hCPC differentiation in different manners. Precisely, genes involved in the differentiation toward cardiomyocytes and smooth muscle cells were upregulated in hCPCs cultured on d-ECM from Pr5, with statistically significant differences (p < 0.05) that were more striking for genes involved in the acquisition of more mature phenotypes, such as sarcomeric actin and smooth muscle actin (Fig. 7).

Real-time polymerase chain reaction analysis of the expression of genes characteristic of cardiomyocytes (MEF2C and ACTC1), endothelial cells (ETS1 and FVIII), and smooth muscle cells (GATA6 and ACTA2). Each value expresses the mean of seven samples (n = 7) ± SD. Asterisks are indicators of the p-value obtained in each comparison as follows: significant (*p ≤ 0.05) and extremely significant (***p ≤ 0.001).
Discussion
Ischemic heart disease is a major cause of death and disabilities in developed countries and the biggest worldwide, as it is the underlying cause of death in approximately one of every seven deaths. Furthermore, according to AHA computations, ∼34% of people who experience a coronary event in a given year will die as a result, and ∼15% who experience a myocardial infarction (MI) will die as a result. In addition, in people who survive the acute stage of an MI, the risk of another MI, sudden death, angina pectoris, heart failure, and stroke is substantial, and they have a 1.5–15 times higher chance of illness and death than the general population.23,24
Although prevention is better than cure, once an ischemic event occurs, the most effective intervention is to replace the damaged myocardium rather than let the damaged tissue repair itself with scar tissue. Therefore, several therapeutic strategies aimed at restoring the integrity of the myocardium and relying on either stem cell transplantation or cardiac tissue engineering have been developed and tested thus far. 25
The prospect of rebuilding the myocardium in its extracellular and cellular compartments would dramatically change the natural course of ischemic heart disease. Nevertheless, while stem cell-based therapies were first introduced about two decades ago, 1 a more complex regenerative approach based on the combination of biomaterials and stem/progenitor cells has recently gained the attention of scientists. In the last decade, numerous biocompatible synthetic materials have emerged as potential extracellular cardiac environment substitutes, 4 although recapitulating ECM structure, organization, and composition remains difficult. The introduction of d-ECM in cardiac regenerative medicine 16 has offered a promising solution to circumvent the issues related to synthetic materials. However, to remove cells from tissues or organs, chemical, physical, or enzymatic methods 26 need to be used, and often a combination of the aforementioned methods is needed to ensure complete cell removal at the cost of ECM partial loss or disruption. Indeed, although several combinations have been tested thus far to thoroughly decellularize the human heart while preserving the ECM, further improvements are needed, as loss of the composition of the ECM27 or the persistence of cellular remnants 28 has been occasionally reported. Furthermore, whether to decellularize the entire heart or to develop engineered heart patches based on naturally occurring ECM remains open to debate.
On one hand, the idea of rebuilding the entire heart starting with a tridimensional biological scaffold as a blueprint is tremendously appealing. However, coaxing administered cells to reach the proper final location and to fully develop into mature, functioning cells remains an ambitious, unachieved goal. Therefore, the construction of cardiac patches as grafts to repair infarcted myocardium and to sustain cell proliferation and differentiation is a short-term and realistic target that may lead to clinical translation.
Many protocols for decellularization of the whole heart have been described so far, while only three protocols for decellularization of pericardium or human myocardium sections can be found in the recent literature. Pursuing the construction of biological patches of human myocardium, we decellularized sections of human cardiac d-ECM according to the three mentioned protocols (Pr1, Pr2, and Pr3) and compared the outcomes in terms of the decellularization efficiency, composition, and structure of the d-ECM scaffolds and suitability for cell repopulation. To the best of our knowledge, this is the first comparison of protocols for the decellularization of human heart, although a number of different protocols have been described to date. In addition, we propose two novel protocols of decellularization both developed by modifying a popular protocol. 16 For the first new protocol, referred to as Pr4, the novelty lies in the usage of sections of human heart, as it is reported for use in whole heart decellularization via antegrade coronary perfusion. In the second new protocol (Pr5), changes were made to the procedure to ensure better and faster decellularization.
The major finding of this study is the substantial improvement in the decellularization procedure in terms of the quality of the scaffold yielded, with few changes to the protocol. Indeed, although affected by the inevitable partial loss of ECM components, Pr5 delivered the most preserved d-ECM in both composition and architecture with the highest potential for myocardium regeneration. Unexpectedly, two of the three previously published protocols were unable to produce a thoroughly acellular ECM suitable for cell repopulation, as they retained donor cell material and were cytotoxic during in vitro testing.
To be of clinical value, the d-ECM should be biocompatible and nonimmunogenic, which implies that both a reduction of nucleic acids below a defined threshold and complete removal of cellular antigens 29 need to be accomplished. Notably, histological analysis revealed the absence of nuclei and cytoplasm in the d-ECM obtained with Pr1, Pr2, Pr4, and Pr5, while cytoplasm remnants were still trapped in d-ECM mesh from Pr3 (Fig. 1). Moreover, Pr5 produced the d-ECM with the lowest residual dsDNA content (Fig. 2), even though a step for the removal of residual nucleic acids was not part of the procedure, as for Pr1, Pr2, and Pr3 (Table 1). The d-ECM obtained with Pr3 instead contained more dsDNA than the currently accepted standard, even after DNase and RNase treatment, and the d-ECM from Pr1 retained amounts of dsDNA that were very close to the mentioned threshold (Fig. 2). On the other hand, while histochemistry (Fig. 3) and immunohistochemistry (Fig. 4) showed that collagen, elastic fibers, and noncollagenous proteins such as fibronectin, laminin, and tenascin were preserved in d-ECM from all protocols, Pr3 produced the d-ECM that retained the highest amount of insoluble collagen (Fig. 5A). Nonetheless, such evidence is reasonably due to the ineffective decellularization procedure that, besides slightly affecting cytoplasmic protein and DNA content, also exerts a milder effect on ECM components. Hence, excluding Pr3 from comparison, Pr2 and Pr5 were the protocols that produced acellular ECM with the highest amounts of collagen. According to the histological analysis, marked differences in the tridimensional architecture of the d-ECM obtained from different protocols also emerged (Figs. 1, 3, and 4). Specifically, myocardium decellularized via Pr4 and Pr5 strictly resembled the native myocardium in which empty spaces substituted cardiomyocytes and were circumscribed by an intricate mesh of collagen and noncollagenous ECM proteins.
Regarding sGAG, it has been previously demonstrated that GAGs interact with several proteins in the ECM and that such interactions protect growth factors, chemokines, and cytokines against proteolysis.21,22 GAGs are also involved in FGF, HGF, VEGF, and PDGF signaling pathways, 22 and thus, preserving GAGs in the d-ECM indirectly preserves growth factors and their biological activity. Although it has been reported that treatment with SDS removes GAGs, 30 in our study, quantitative measurements demonstrated that d-ECM obtained with all five protocols retained sGAG (Fig. 5B). Interestingly, even though the d-ECM from Pr5 and Pr1 contained higher sGAG contents, the amounts were comparable in the d-ECM from all protocols, with the only exception of Pr4, which retained lower sGAG content. Because Pr4 requires 3 days longer than all other protocols, we can infer that this difference may be due to the duration of the procedure and to the time the sections were kept in solution.
As consequence of GAG resistance to the decellularization procedure, the d-ECM obtained with all five protocols also retained growth factors. The d-ECM contained growth factors involved in biological processes such as cell proliferation and differentiation, including EGF, FGF,31–34 and growth factors known to modulate the maintenance of stemness and stem cell proliferation and migration, including IGF and SCF.35–37 Intriguingly, receptors for growth factors, such as EGF, PDGF, SCF, and VEGF, were still stored in the d-ECM obtained with all protocols with the exception of the d-ECM from Pr5 (Fig. 6). Because growth factor receptors are proteins located on cell membranes, their absence from the d-ECM obtained with Pr5 should be considered as evidence that effective removal of cell membrane remnants occurred with Pr5, but not with the other four protocols. It is noteworthy that sometimes in d-ECMs from Pr1, Pr3, and Pr4, significantly higher levels of growth factors (Supplementary Fig. S1), such as EGF, M-CSF, PDGF-AA, PDGF-AB, and VEGF, are paralleled by significantly higher levels of their respective receptors (Supplementary Fig. S2).
d-ECM holds great promise as a powerful regenerative tool due to its versatility. Specifically, potential clinical applications include either the implantation of acellular d-ECM to deliver physiological stimuli that can boost regeneration through activation of resident primitive cells or the construction of tailored patches recellularized with patient-derived stem or progenitor cells. However, the assessment of effects exerted by the d-ECM on either resident or seeded cells is the most critical step before considering preclinical and clinical translation. Notably, tests of in vitro recellularization of d-ECM with hCPCs showed that the d-ECMs obtained via Pr1 and Pr3 were unsuitable for cell repopulation, as seeded cells detached and died within 48 h, possibly due to the toxic effect of cell remnants in the d-ECM. In contrast, hCPCs successfully repopulated in vitro d-ECM sections obtained with Pr2, Pr4, and Pr5. However, cell viability on d-ECMs obtained from the three aforementioned protocols differed significantly and the d-ECM obtained from Pr5 offered the most favorable environment for hCPC survival in vitro (Supplementary Fig. S3). Gene expression profile revealed that hCPCs cultured in the d-ECM scaffolds expressed genes involved in the differentiation of cardiomyocytes, endothelial cells, and smooth muscle cells, although genes characteristic of a precursor state, rather than a progenitor state, were upregulated in hCPCs cultured on d-ECM from Pr5 (Fig. 7). It is not surprising that the microenvironment provided by the d-ECM from Pr5, preserving biological signals and tridimensional architecture while thoroughly removing cell remnants, better supported the terminal differentiation of CPCs toward myocardium and smooth muscle cell lineages. Notably, previous studies demonstrated that, when cultured on culture dishes, hCPCs isolated from explanted hearts failed to reach the more mature phenotype of precursor cells and expressed mostly markers of a progenitor state, 38 while the presence of the d-ECM prompted progenitor cells to further proceed to differentiate toward precursor and mature phenotypes. It is also noteworthy that Pr4 and Pr5 decellularization procedures are the only two procedures that are based only on two chemicals, making them easier to perform and safer for recellularization. 39 In addition, comparing the Pr4 and Pr5 protocols, the latter is much shorter and more effective, as the d-ECMs obtained exhibited better-preserved tridimensional architectures and contained higher amounts of collagen and sGAG but lower amounts of dsDNA. Altogether, evidence collected in our comparative study shows that Pr5 is the simplest and most effective protocol for the decellularization of human myocardium sections and the protocol that delivered the best preserved in terms of composition and tridimensional architecture and the best performing d-ECM in terms of ability to support hCPC differentiation. Such apparent inconsistency is likely due to the combined effect exerted by Triton X-100 and SDS on cellular compartments. In fact, Triton X-100 is a nonionic, nondenaturing, mild detergent that disrupts lipid–lipid and lipid–protein interactions while leaving protein–protein interactions intact. Therefore, solubilizing the membranes exposes tissues to the action of SDS that removes nuclear remnants and cytoplasmic proteins and emulsifies fats. 27 In addition, previous studies suggested that rinsing with Triton X-100 decreases SDS content in decellularized heart, 16 although it has been demonstrated that a 6-h wash after liver decellularization with 1% SDS is enough to highly reduce the residual SDS to undetectable levels by gas chromatography quantification 40 and that no measurable amounts of residual SDS can be detected in hearts that are decellularized with 1% SDS. 41
Conclusion
While comparisons of decellularization procedures for rat heart 42 or other tissues and organs43–47 have been published in the last decade, to the best of our knowledge, this is the first comparison of protocols for the decellularization of human myocardium. The importance of this comparison pertains to scientific data that can be acquired while saving time and resources and without having to test several protocols in search of the most effective. In summary, of the five decellularization protocols tested, only three protocols yielded acellular d-ECM scaffolds with well-preserved structures. Among these three, we developed a new decellularization protocol, which, although based on a previously described protocol, introduces changes that ensure better results and performance during in vitro recellularization testing. Partial loss of ECM components is inevitable but still acceptable considering that the best alternative bioengineered scaffolds consist primarily of synthetic polymers that partially recapitulate the composition of the ECM through the incorporation of ECM protein fragments. In addition, naturally occurring ECM has considerable biocompatibility, and, as such, holds the greatest potential for engrafting in host tissue while avoiding an immune response. Altogether, the newly tested Pr5 delivers d-ECMs that better meet the recently proposed criteria for cell-free ECM scaffolds 29 and better support hCPC survival and differentiation in vitro, and this protocol is worth improving and testing in vivo.
Footnotes
Acknowledgment
This work was supported by the 20123E8FH4_002 research grant, funded by the Italian Ministry of Education, University and Research.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.