
Abstract
Extracellular matrix (ECM) is an essential component of the tissue microenvironment, actively shaping cellular behavior. In vitro culture systems are often poor in ECM constituents, thus not allowing for naturally occurring cell–ECM interactions. This study reports on a straightforward and efficient method for the generation of ECM scaffolds from lung tissue and its subsequent in vitro application using primary lung cells. Mouse lung tissue was subjected to decellularization with 0.2% sodium dodecyl sulfate, hypotonic solutions, and DNase. Resultant ECM scaffolds were devoid of cells and DNA, whereas lung ECM architecture of alveolar region and blood and airway networks were preserved. Scaffolds were predominantly composed of core ECM and ECM-associated proteins such as collagens I-IV, nephronectin, heparan sulfate proteoglycan core protein, and lysyl oxidase homolog 1, among others. When homogenized and applied as coating substrate, ECM supported the attachment of lung fibroblasts (LFs) in a dose-dependent manner. After ECM characterization and biocompatibility tests, a novel in vitro platform for three-dimensional (3D) matrix repopulation that permits live imaging of cell–ECM interactions was established. Using this system, LFs colonized the ECM scaffolds, displaying a close-to-native morphology in intimate interaction with the ECM fibers, and showed nuclear translocation of the mechanosensor yes-associated protein (YAP), when compared with cells cultured in two dimensions. In conclusion, we developed a 3D-like culture system, by combining an efficient decellularization method with a live-imaging culture platform, to replicate in vitro native lung cell–ECM crosstalk. This is a valuable system that can be easily applied to other organs for ECM-related drug screening, disease modeling, and basic mechanistic studies.
Introduction
E
Several lung diseases involve dramatic ECM remodeling, which results in excessive deposition of ECM components and, subsequently, in heterogeneous ECM stiffening, compromising organ function. In fact, the feedback loop of reciprocal interaction between pathological ECM and fibroblasts greatly impacts pulmonary disease progression. In idiopathic pulmonary fibrosis (IPF), a restrictive lung disease associated with high morbidity and mortality, components of ECM are highly degraded by matrix metalloproteinases (MMPs), making MMPs promising targets for future therapy. 6 Other potential targets for therapy of IPF are the mechanosensors yes-associated protein (YAP) and TAZ, as RNAi knockdown of YAP/TAZ has been shown to attenuate the fibrotic behavior of IPF-derived LFs. 7 Currently, no well-characterized murine model exists for IPF.8,9 Three-dimensional (3D) in vitro models based on lung natural ECM scaffolds are promising tools to functionally assess target proteins on patients' cells in a 3D native-like context.
However, generation of robust and consistent in vitro models to study the interaction of native lung ECM with the various lung cell types is yet challenging. Isolation of ECM from native lung tissue involves the use of detergent-based protocols, which impact the biochemical structural properties of the resultant ECM scaffold. The use of ionic detergents during the decellularization process removes structural ECM proteins, compromises the integrity of basal membranes, and alters the resultant bioactive properties of lung ECM scaffold.10,11 Therefore, the concentration of these detergents should be minimized. A high pH during decellularization, commonly employed to remove residual DNA, eliminates ECM-bound glycosaminoglycans, which retain growth factors that constitute the bioactivity of the ECM. 12 Each tailored protocol differently impacts the structural and biochemical properties of the ECM and, subsequently, the biological effect of the generated ECM on cells during subsequent in vitro seeding experiments. 13 Although these evidences demonstrate the greatest importance of high-throughput biochemical characterization of the decellularized ECM, studies often provide limited evidence of ECM preservation.11,14,15 To ensure homogeneous and effective removal of cellular material, the tissue must be uniformly permeated by decellularization solutions. 14 In line with this, advanced perfusion systems, not commonly available to all researches, are often required to decellularize lung tissue.16–18
Here, we present a straightforward decellularization protocol to prepare ECM scaffolds from mouse lungs with preserved microstructure, composition and with negligible contamination of DNA. Protein identification and quantification by mass spectrometry showed that ECM scaffolds retained a diversity of ECM components with the marginal presence of cellular proteins. We established an in vitro platform for matrix repopulation that permits straightforward visualization of cells by confocal and electron microscopy. In a proof-of-principle experiment, lung ECM scaffolds were shown to promote attachment, cell spreading, and proliferation of primary LFs. In addition, preferential nuclear location of the YAP in LFs further demonstrated cellular mechano-responsiveness to the ECM. This work details the generation of an in vitro 3D-like culture system that is relevant for researchers aiming at replicating the in vivo cell–ECM crosstalk for understanding basic biological mechanisms, drug screening, and disease modeling.
Materials and Methods
Decellularization of mouse lung
The decellularization protocol is an adaptation (Fig. 1A) of a previously established protocol for mouse heart. 19 Adult mice C57BL/6 (6–9 weeks of age) were euthanized by carbon dioxide. A median sternotomy was performed in a sterile fashion, and lungs were excised and cleaned with phosphate-buffered saline (PBS). The left lobe was divided in 10 parts (Fig. 1B). Samples were embedded in optimum cutting temperature (OCT, Cryomatrix 6769006; Thermo Scientific), cryopreserved in liquid nitrogen-cooled isopentane (2-methyl butane; Sigma), and stored at −80°C up to 30 days. For decellularization, samples were defrosted, washed for 15 min in PBS under agitation (165 rpm), and incubated for 18 h in hypotonic buffer (10 mM Trizma base [Sigma], 0.1% EDTA [Sigma-Aldrich]). Next, the fragments were washed three times with MilliQ water (1 h per wash), followed by incubation in 0.2% sodium dodecyl sulfate (SDS; Sigma-Aldrich) for 24 h. Samples were then washed twice for 20 min in 10 mM Trizma base, followed by a long wash overnight in the same buffer. For DNA removal, samples were incubated for 6 h in a 50 U/mL DNase (Sigma) solution, which was refreshed after 3 h of treatment. After incubation with DNase, tissue fragments were washed three times in MilliQ water (20 min per wash) at room temperature. All incubation steps were performed in an incubated shaker at 25°C except for the DNase treatment, which was performed at 37°C. Decellularization solutions were adjusted to pH 7.8. To prevent contamination, all incubation and washing solutions contained a final concentration of 2.5 μg/mL of fungizone (Thermo Fisher Scientific, 15290018) and 10 μg/mL of gentamicin (Thermo Fisher Scientific, 15750060).

Effective adult lung decellularization. Schematic representation of the decellularization procedure
Histology and immunofluorescence
Decellularized and native lung tissues were fixed in 4% paraformaldehyde (PFA) for 5 h followed by washing three times with PBS for 10 min and then embedded in OCT for immunostaining and in paraffin for histological staining. Thin sections (3 μm) were prepared for hematoxylin and eosin (HE) and Masson's trichrome (MT) staining as previously described 20 and immunostaining. For immunostaining, sections were permeabilized with 0.2% Triton in PBS for 5 min and blocked in 4% fetal bovine serum (FBS)/1% bovine serum albumin (BSA) in PBS for 1 h. Sections were then incubated with primary antibodies in 4% FBS (Lonza)/1% BSA (Sigma) in PBS:Collagen IV (goat, 1:50, 8 μg/mL; Millipore, AB769), fibronectin (rabbit, 1:400, 1.5 μg/mL; Sigma-Aldrich, F-3648), or laminin (rabbit, 1:400, 1.25 μg/mL; Sigma-Aldrich, L9393). Secondary antibodies used were: Alexa Fluor 555-conjugated donkey anti-rabbit (A-31572, 1:500, 4 μg/mL; Thermo Fisher Scientific, A21429) and Alexa Fluor 568-conjugated donkey anti-goat (1:500, 4 μg/mL; Thermo Fisher Scientific, A-11057). Sections were incubated with secondary antibodies for 1 h at room temperature and counterstained with DAPI (dilution 1:1000; Thermo Fisher Scientific, D1306). The sections were visualized on an inverted microscope (LSM 800; Zeiss). For immunostaining of collagen I and nephronectin, native and decellularized adult lungs were formalin fixed for 2.5 h at room temperature and processed for paraffin embedding. Overall, 5 μm sections of the lung samples were subjected to a heat-induced epitope retrieval and blocked with 1.5% normal donkey serum (NDS; Fisher Scientific, NC9624464) in PBS for 1 h. Collagen I (1:300, 3.3 μg/mL; Abcam, ab34710) and nephronectin (1:100, 5 μg/mL; Abcam, ab64419) were diluted in 1.5% NDS, applied onto the samples, and incubated overnight at 4°C. Then, the tissue sections were incubated for 1 h with the Alexa Fluor 555-conjugated donkey anti-rabbit secondary (1:400, 5 μg/mL; Invitrogen, A-31572) and Hoechst 33342 (2 μM; Thermo Fisher Scientific, 62249) in 1.5% NDS at room temperature. Sudan Black B (Electron Microscopy Sciences, 21610) treatment was performed for 20 min, protected from light, to reduce tissue autofluorescence. Tissue-stained sections were mounted with ProLong™ Gold antifade mounting medium (Thermo Fisher Scientific, P36930), and images were acquired by using the Zeiss inverted microscope.
Extraction and quantification of DNA
Genomic DNA was isolated in parallel from decellularized and native lung tissue by using PureLink Genomic DNA Mini Kit (Invitrogen) according to the manufacturer's instructions. DNA was labeled by using Qubit dsDNA Assay Kit (Invitrogen) and measured on Qubit 2.0 fluorometer (Invitrogen). Assay was performed on three individual samples from decellularized and from native lungs.
Quantification of tissue density
Sections of native and decellularized lung samples were stained with HE and photographed by using Leica CTR 5000 microscope. Then, binary images were generated by using ImageJ. 21 The default auto-thresholding method was used to generate the binary images. Tissue density was quantified by calculating the percentage of the area occupied by tissue regarding the total area of each binary image. Quantification was performed on three individual sections each from native and decellularized lung.
Scanning electron microscopy
Native and decellularized lung tissue was sectioned into cubic explants of ∼2 × 2 × 2 mm and processed for scanning electron microscopy (SEM). Decellularized ECMs with LFs were processed while they still adhered to the bottoms of Petri dishes with a thin bottom for imaging purposes (Ibidi). 22 All samples were fixed in 3% glutaraldehyde in 100 mM cacodylate buffer for 2 h and then washed in cacodylate buffer (100 mM sodium cacodylate in distilled water) for 15 min. The samples were dehydrated in 30%, 50%, 70%, 80%, and 96% ethanol gradient (15 min per each concentration). Subsequently, the samples were dried at critical point dryer CPD 030 (Balzers Union Limited), sputtered with gold in a sputter coater SCD 030 (Balzers Union Limited), and observed by means of a scanning electron microscope (Vega, Tescan).
Transmission electron microscopy
Native and decellularized lung tissue was sectioned into cubic explants of 2 × 2 × 2 mm, fixed in 3% glutaraldehyde in 100 mM cacodylate buffer for 2 h, and finally washed in cacodylate buffer (100 mM sodium cacodylate in distilled water) for 1 h. Samples were dehydrated in 70%, 96%, and 100% ethanol grade and then in 100% acetone at 4°C, 40 min for each step. Subsequently, the samples were embedded in epoxy resin Durcupan (Durcupan ACM; Sigma). Ultrathin sections were prepared on ultramicrotome (LKB 8802A) and observed by means of transmission electron microscopy (Morgagni 268D TEM; FEI).
Mass spectrometry analysis
Four samples (each from different mice and from different decellularization experiments) were processed for liquid chromatography-mass spectrometry (LC-MS) analysis. Detailed description of the method is available in Supplementary Methods (Supplementary Data are available online at
Isolation of primary mouse LFs
Lungs from ICR mice (6–7 weeks old) were excised, washed with PBS, and minced by using a scalpel. The sorting strategy was adapted from a previously published protocol. 23 A detailed protocol of cell isolation and sorting strategy is outlined in Supplementary Methods.
Cell culture on ECM slices
Decellularized lung tissue was embedded in OCT compound (Tissue-Tek) and sectioned on cryomicrotome into 25 μm-thick, slices, which were then left to adhere to the bottom of an Ibidi dish for 20 min at 37°C. The remaining OCT was removed by three washes with PBS. LFs were seeded at the top of ECM slices that adhered to Ibidi dishes at a density of 50,000 cells/cm2. The samples were fixed in 4% PFA and further analyzed after 6 days of culture.
Immunostaining of repopulated ECM slices
Repopulated ECM slices were fixed in 3.7% formaldehyde for 15 min, permeabilized by using 0.5% Triton X-100, and blocked in 3% BSA in PBS for 60 min. The samples were stained by using different primary antibody cocktails overnight at 4°C. The primary antibodies used were: Collagen IV (rabbit, dilution 1:200, 5 μg/mL; ab6586), MMP-2 (rabbit, dilution 1:200, 5 μg/mL; ab37150), and MMP-9 (rabbit, dilution 1:500, 2 μg/mL; ab38898) from Abcam, and YAP (mouse, dilution 1:200, 0.5 μg/mL; Santa Cruz Biotechnology, sc-101199). After a washing step with PBS, the incubation with secondary antibodies was performed for 1 h at room temperature. The secondary antibodies applied were: Alexa Fluor 488-conjugated anti-rabbit (dilution 1:600, 3.3 μg/mL; A-11008), Alexa Fluor 568-conjugated anti-rabbit (dilution 1:600, 3.3 μg/mL; A-11036), and Alexa Fluor 568-conjugated goat anti-mouse (dilution 1:600, 3.3 μg/mL; A-11031) antibodies (all from Thermo Fisher Scientific). Phalloidin-Rhodamine (dilution 1:400; Thermo Fisher Scientific, R415) was added together with secondary antibodies and incubated for 1 h at room temperature. Nuclei were counterstained with DAPI for 5 min, and the resulting samples were imaged with a scanning confocal microscope (Zeiss LSM 800).
Staining of repopulated ECM slices with EdU
Cryopreserved ECM thick slices previously seeded with LFs for 6 days were stained with Click-iT™ EdU Alexa Fluor 594 HCS Assay (Invitrogen) as follows: 20 μM EdU was added to culture media for 1 h at 37°C and then fixed in 3.7% formaldehyde for 15 min. Samples were permeabilized by using 0.5% Triton X-100 in PBS. Click-iT reaction cocktail containing Alexa Fluor 549 azide was added and left for 30 min at room temperature, protected from light. Samples were blocked in 3% BSA in PBS and stained with primary antibody for collagen IV (rabbit, dilution 1:200, 5 μg/mL; Abcam, ab6586) overnight at 4°C. Then, the samples were washed, incubated with Alexa Fluor 488-conjugated anti-rabbit antibody (goat, dilution 1:600, 3.3 μg/mL; A-11008), and counterstained with DAPI (dilution 1:1000).
Homogenization of lung ECM
For the lung ECM to be applied as coating, the samples were first mechanically homogenized. After the decellularization process, each explant of decellularized lung was separately immersed in 200 μL of sterile PBS and mechanically disrupted by ceramic beads (MagNA Lyser Green Beads; Roche) by using a homogenizator (MagNA Lyser; Roche). Samples were homogenized three times for 25 s at 6000 rpm and cooled on ice for 1 m between each homogenization round to prevent overheating. Protein concentration was measured with DC Assay according to the manufacturer's instructions (Bio-Rad). For each culture experiment, fresh ECM was isolated and homogenized before application.
Attachment assay
A 96-well plate was coated with homogenized lung ECM diluted in PBS and left overnight at 37°C. Three concentrations of homogenized ECM were used: 10, 100, and 1000 ng/mL, as well as PBS alone as a control. The volume of coating solution was (600 μL/cm2). LFs were seeded at a number of 30,000 cells/cm2 and left to attach for 30 min. Subsequently, the medium was removed and cells were fixed in 3.7% formaldehyde for 20 min. After being washed twice with PBS, cells were incubated for 60 min in 0.1% crystal violet solution followed by three washes in PBS. Crystal violet accumulated in cells was then dissolved in 33% acetic acid, and absorbance was measured at 590 nm with a multi-mode reader (Synergy HTX; BioTek). The assay was performed in three independent experiments by using technical triplicates in each.
Statistical analysis
Results are shown as averages ± standard mean error. The F-test for variance was performed to test variance of results followed by the two-tailed t-test to determine whether any group was significantly different from control (p < 0.05).
Results
Decellularization of lung explants
Lung ECM was obtained by subjecting alveolar tissue fragments to decellularization (Fig. 1A). Cell removal was attained by three main steps: (1) cell lysis by osmotic shock (hypotonic buffer), solubilization of lipid–protein, DNA–protein, and protein–protein interactions (using a low concentration—0.2% of SDS), and nuclear material removal (DNase treatment) (Fig. 1A). During decellularization, the tissue progressively became whiter and in thinner regions transparent, indicating cellular removal (Fig. 1B). Clearance of nuclei was evidently showed by absence of DAPI staining. Quantification of DNA isolated from decellularized and native tissue further confirmed removal of >99% of DNA (from 261.7 ± 30.14 ng/mg to 1.757 ± 0.211 ng/mg, respectively) (Fig. 1C). HE staining showed normal distribution of cells in native mouse lung tissue, whereas decellularization effectively removed cellular components while preserving the typical porous alveolar structure (Fig. 1D). MT staining evidenced the preservation of collagen network (blue-stained) after decellularization (Fig. 1D). The removal of cellular components was also evidently showed by decreased tissue density of the explants. Native tissue in the alveolar compartment exhibited average 35.18% ± 1.16% of tissue covered area whereas the decellularized tissue exhibited average 22.3% ± 2.15%, resulting in a 12.9% decrease of tissue density after the decellularization procedure (Fig. 1E).
Proteomic analysis of lung ECM
A precise combination of ECM proteins defines the cellular microenvironment of a specific tissue in normalcy and in a pathological state. To obtain the proteomic signature of lung ECM, we subjected decellularized scaffolds to proteomic analysis by mass spectrometry. Functional clustering of identified proteins, based on in silico Matrisome database (
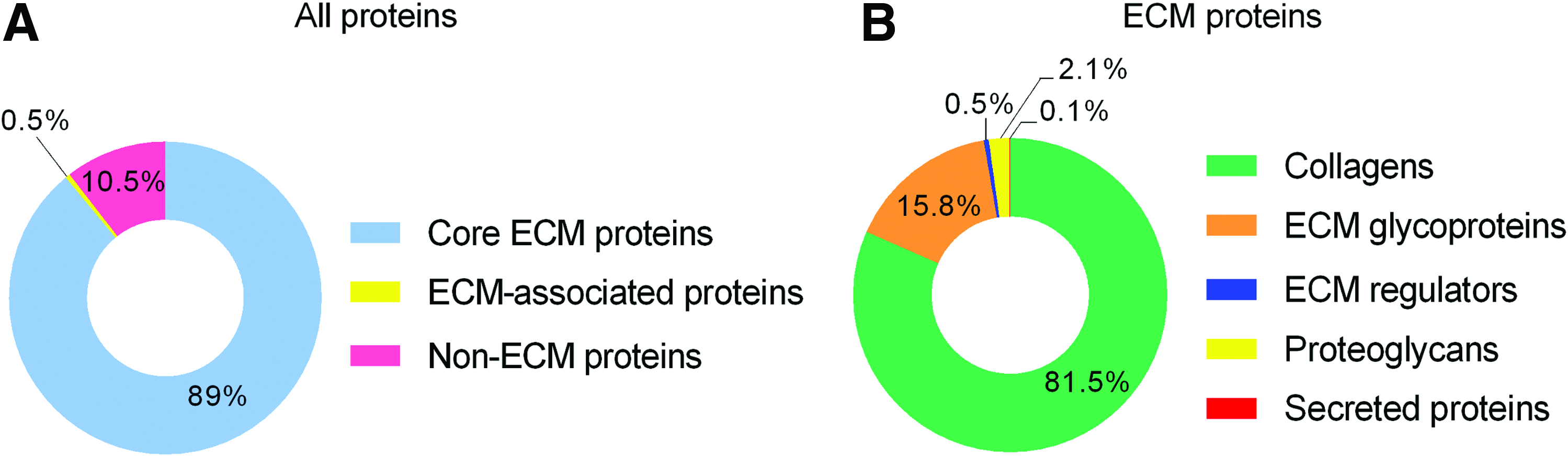
High-throughput proteomic analysis of decellularized mouse lung. Doughnut chart shows coverage percentage of the total amount of protein identified by mass spectrometric analysis and classified as core ECM proteins, ECM-associated proteins, and non-ECM proteins
Collagens, which are the critical ECM structural components, were identified as the most abundant group in lung ECM scaffolds, predominantly represented by collagen I, III, and IV (Fig. 2B). The second most represented group of proteins were adhesive glycoproteins such as laminin, fibrillin, and fibronectin. We repeatedly identified nephronectin, an alpha-8 and beta-1 integrin-binding ECM protein identified in various organs including kidney and lung,26,27 as the most abundant glycoprotein present in the decellularized lung (Table 1).
The most abundant proteins within categories of core ECM proteins, ECM-associated proteins, and non-ECM proteins in decellularized mouse lung identified by mass spectrometric analysis.
ECM, extracellular matrix; ppm, parts per million.
Proteoglycans were also abundant in the decellularized lung as shown by the identification of their core protein, namely, heparan sulfate proteoglycan core protein, the third most represented group of proteins in ECM scaffold. Heparan sulfate is an essential regulator of cell-growth factor interaction and creates local gradients by the immobilization of soluble bioactive proteins. Of the total protein content, 0.5% were identified as ECM regulators, which are proteins responsible for crosslinking of ECM proteins and are essential for regulation of ECM topography. The most abundant ECM regulators present in the scaffold are transglutaminase 2 and lysyl oxidases. Of note, the secreted protein multiple epidermal growth factor-like domains protein 6 (MEGF6), whose function is yet unknown, was detected in homogenate as relatively abundant.
Transforming growth factor β (TGF-β) signaling is active during lung development and actively participates in tissue homeostasis and repair in adult lung. 28 In homeostatic conditions, TGF-β is immobilized in the ECM in complex with latency-associated peptides and latent TGF-β-binding proteins (LTBPs). 29 We identified latent TGF-β-binding protein 1 (LTBP-1) and latent TGF-β-binding protein 4 (LTBP-4) in lung ECM scaffolds. It has been described that LTBP-4 associates with fibrillin 1 and LTBP-1 associates with fibronectin, 30 which were also detected in lung ECM samples.
Quantitatively, the majority of non-ECM proteins in the scaffold are cytoskeletal proteins actin and tubulin and also residual myosin proteins from lung smooth muscle cells. We detected transforming growth factor beta-1-induced transcript 1 protein (TGFB1I1), which is a part of the focal adhesion protein complex. 31 Among these non-ECM proteins, we also detected integrins beta-1 and beta-3. The presence of cytoskeletal proteins and integrins suggests that after decellularization a fraction of cell adhesion complexes remained in the ECM scaffold. A complete list of all identified proteins is provided in Supplementary Table S1.
Having assessed the biochemical composition, the effect of decellularization on the distribution of abundant ECM components was assessed by immunofluorescence. Collagen IV and laminin are major components of the basal laminae that constitutes a substantial fraction of the lung ECM. Both collagen IV and laminin remained similarly expressed and distributed, showing a continuous network pattern, in scaffolds when compared with the native tissue (Fig. 3A, B, F, G). In addition, fibronectin and nephronectin, ECM glycoproteins that bind to integrins to enable ECM–cell interactions, were also retained in the ECM scaffold with a distribution similar to the native tissue (Fig. 3C, D, H, I). Collagen I, the main ECM molecule that contributes to tissue structure and stiffness, was equally present and distributed on nonmanipulated lung tissue and decellularized scaffolds (Fig. 3E, J). These data indicate that the decellularization procedure generates acellular lung scaffolds with preserved composition and localization of main structural and adhesive ECM proteins.

Main structural ECM proteins network is preserved after decellularization. Representative images of immunofluorescence for collagen IV
Architecture and microstructure of acellular lung ECM scaffolds
To observe the impact of decellularization on ECM microarchitecture, SEM was used to visualize native tissue and decellularized ECM scaffolds. SEM images of native lung tissue showed the alveolo-capillary network lined with epithelial and endothelial cells (Fig. 4A) as well as airway and vessels (Fig. 4B) lined with airway epithelial (Fig. 4C) and endothelial cells (Fig. 4D), respectively. In contrast, in the respective decellularized regions, only denuded basal membranes without cells were observed (Fig. 4E–H). Microtopography of vascular basal membrane in decellularized sample is smooth (Fig. 4H) compared with the more irregular basal membrane of the decellularized airway (Fig. 4G). TEM images of decellularized alveolar region further confirmed preserved alveolar walls, consisting of thin basal membranes separating the alveolar space from the surrounding capillary network (Fig. 4I). The wall of the airway was also well conserved on the decellularized sample as demonstrated by the presence of collagen IV, elastin, and fibrillin (Fig. 4J). Both microscopic techniques demonstrated that decellularization efficiently retained architectural and structural features, namely the basal membrane of alveolar and bronchiolar regions, without visible cellular debris.
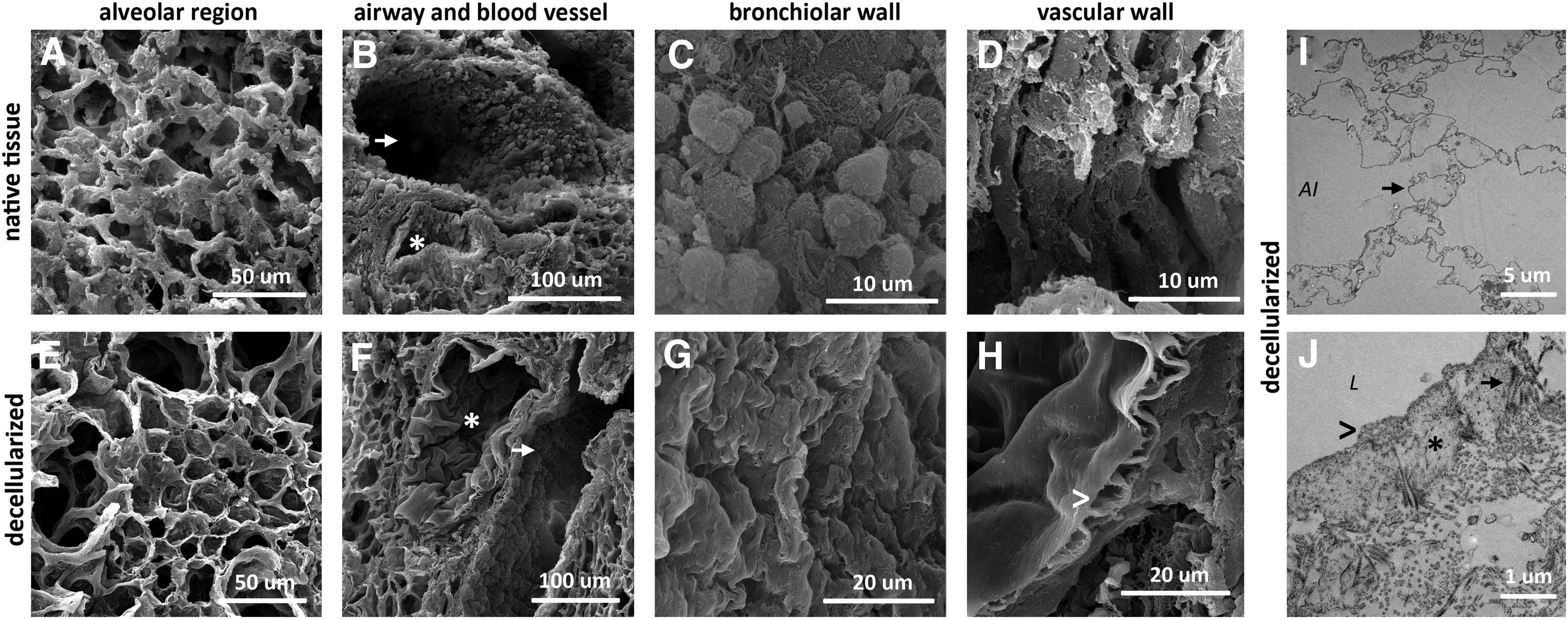
Morphology of different lung anatomical regions is preserved after decellularization. Representative images of scanning electron microscopy showing the structure of the lung alveolar compartment of native lung tissue
Bioactivity of homogenized ECM
One important function of ECM is to facilitate and support cell attachment. To assess whether decellularized lung ECM retained this capacity independently on structural cues, a two-dimensional (2D) cell culture setup was implemented, in which mechanically homogenized ECM was used to coat cell culture dish surfaces to improve cell attachment. Hence, primary mouse LFs were seeded directly on pristine tissue culture polystyrene (TCPS) dishes or on TCPS dishes coated with mechanically homogenized ECM and harvested 30 min after seeding to quantify cell attachment. A trend of dose-dependent increase in the number of cells attached to ECM-coated culture surfaces was observed when compared with uncoated TCPS (Fig. 5A). ECM concentrations of 100 and 1000 ng/mL significantly improved cell attachment (1.27-fold ± 0.024-fold and 1.37-fold ± 0.054-fold, respectively) in concentrations that are notably lower than those typically used for coating cell culture dishes with commercially available components of ECM such as collagen (30 μg/mL) and fibronectin (10 μg/cm2). 32
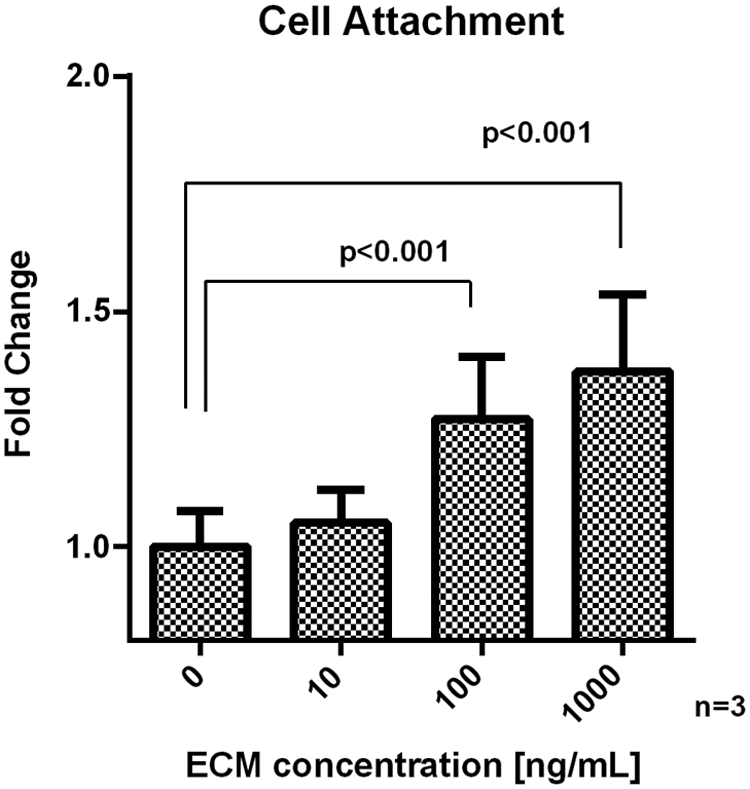
ECM-coated surface promotes attachment of LFs. Cells were seeded on cell culture plastic dishes coated with an increasing concentration of mechanically homogenized lung ECM ranging from 0 to 1000 ng/mL. Thirty minutes after seeding, the unattached cells were washed away and the attached cells were quantified by using Crystal violet assay. Data are expressed as mean ± standard error of the mean. Mean values were compared between pairs of groups by the two-sample t-test. LFs, lung fibroblasts.
Cell response to lung ECM scaffolds
Having established the effect of ECM homogenate, uncoupled from the structural cues, we aimed at investigating the interaction of LFs with ECM scaffolds in 3D conditions. Repopulation of large 3D scaffolds of decellularized matrices often results in uneven cell distribution, cell clustering, and decreased cell viability, and it requires dynamic culture in bioreactors to support oxygenation, nutrient availability, and waste dispersal. 33 In addition, large samples are difficult to observe by standard confocal microscopes. To bypass these limitations, we have adopted a 3D-like format of cell culture conditions in which ECM slices are adhered to culture plastic for cell culture purposes. A similar approach has been previously used with ECM slices of 2 mm, 1 mm, 600 μm, and 100 μm15,18,34,35; however, the thickness of these specimens limits confocal imaging due to tissue scattering and low penetrance of light. Decellularized lung ECM was cryosectioned at different thicknesses (ranging from 20 to 60 μm). We observed that 25 μm-thick slices adhered well to Ibidi culture plastic and exposed important anatomic structures of the lung ECM, which could be well visualized by using standard confocal microscopy and further used for 3D image reconstruction.
To allow adequate repopulation of scaffolds, LFs were cultured for 6 days on decellularized ECM before assessment of cell distribution and morphology. Cells were highly spread and established cell–cell contact, forming multicellular homogenous networks in close contact with the ECM meshwork (Fig. 6A, left). Moreover, cells were able to proliferate in the ECM as visualized by using EdU Click-IT proliferation assay (Fig. 6A, right), suggesting active repopulation of the scaffolds. The morphology of cells growing on scaffold was elongated, accompanied the microstructure of ECM meshwork, and interacted with the ECM via filopodia (Fig. 6B).
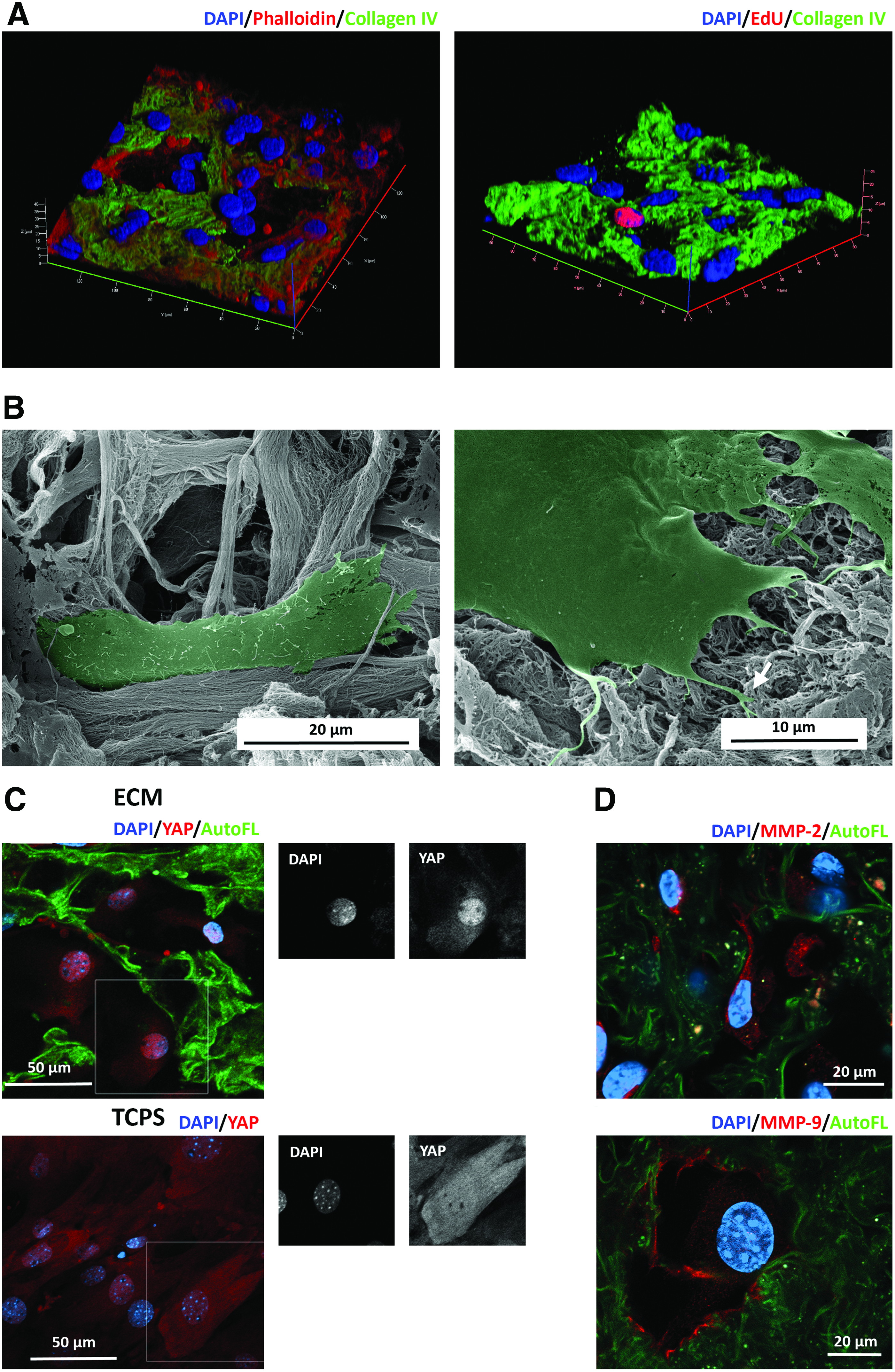
Colonization of ECM scaffolds with LFs. Twenty-five-micrometer-thick slices of acellular ECM scaffolds were seeded with LFs and analyzed by confocal and electron microscopy at day 6 of culture
Cell–ECM interactions are regulated at the biochemical, structural, and mechanical level. Transcriptional effectors of Hippo pathway, YAP and TAZ, which are highly regulated by matrix stiffness, coordinate fibroblast activation and matrix synthesis. 7 We show that YAP protein was expressed by LFs in vitro and that its intracellular localization was affected by the extracellular environment. Decellularized lung scaffolds promoted nuclear YAP localization, whereas cells on TCPS showed both nuclear and cytoplasmic localization of YAP (Fig. 6C).
Cells not only sense the ECM but also modulate the ECM in a feedback manner. Indeed, breakdown and remodeling of ECM is facilitated by matrix metalloproteinases MMP-2 and MMP-9 under a variety of physiological and pathological conditions. 8 In our 3D-like settings, MMP-2 was expressed throughout the cytoplasm of LFs inside the ECM scaffold, whereas MMP-9 was mostly localized at the margins of the cell cytoplasm, in regions of close cell–ECM contact (Fig. 6D). These findings collectively show that the generated bioscaffolds preserve native ECM biochemical and structural properties and represent a practical and adequate in vitro setup to study cell–ECM interactions in a native-like context.
Discussion
Structural and biochemical alterations of lung ECM are commonly reported in several diseases, including IPF, 36 asthma,37,38 and chronic obstructive pulmonary disease. 39 In line with this, lung ECM has been recently suggested to have an impact in disease initiation and progression, making ECM a prospective target for therapy. 40 Decellularized scaffolds may retain tissue architecture, composition, and spatial distribution of ECM proteins, thus constituting native-like environments for cells when cultured ex vivo. Through the use of decellularized ECM, recent studies reported that differences in structure and composition of lung ECM throughout aging 41 and in pathological scenarios 5 differentially have an impact on cells as a feedback loop. These new data, together with the technical advancement in decellularization, pave the way for the use of ECM scaffolds as in vitro models to dissect pathologic processes and identify therapeutic targets for the treatment of pulmonary diseases.
Decellularization protocols must be adjusted to the density and organization of the targeted tissue, and detergent concentrations should be minimized to reduce alterations to the composition and architecture of the ECM. 42 Here, we present a method for lung tissue decellularization by using a low concentration of SDS (0.2%), which does not require any other specialized methods or equipment and preserves the composition and 3D structure of alveoli and the airway/blood vessel network. It has been previously shown that SDS-based decellularization protocol has a negative effect on the integrity of basal membranes of the porcine urinary bladder. 10 Decellularization of lung tissue by 0.5% SDS had compromising effects on small vessels and alveolar septa. 16 However, we provide evidence that in case of lung tissue decellularization with our low-SDS protocol both lung tissue architecture and basal membrane integrity are left intact. Moreover, the use of a nonperfusion system for decellularization permits the application of this technique in nonintact tissues, as clinically relevant tissue biopsies that can be further used for drug screening studies and/or dissection of disease mechanisms. We observed preservation not only of important ECM molecules, such as collagen I, heparan sulfate proteoglycan-binding protein, and nephronectin, but also of ECM-associated factors, such as growth factors and MMPs. Residual DNA in the ECM scaffolds may compromise biocompatibility because it elicits an inflammatory reaction after animals' transplantation. 43 ECM scaffolds prepared by our protocol were devoid of >99% of DNA compared with native lung tissue.
Mass spectrometry provides sensitive compositional characterization of the ECM. We identified a wide spectrum of ECM proteins and also several residual cellular proteins, which constitute a minor proportion of the sample. Of note, proteins with complex fibrillar organization and crosslinking, such as collagen I and elastin, are highly insoluble and even after trypsin digestion their peptides are underrepresented in the LC-MS analysis.44,45 Out of the ECM proteins identified, the proteins affecting cellular responses during subsequent recellularization of scaffolds are of particular interest. Fundamental processes in cells are regulated by discoidin domain receptors, cell surface tyrosine kinases, which are activated by binding to collagens I-IV. 46 Collagen IV and laminins, which are among the most abundant components of lung ECM bioscaffolds, promote cell adhesion by binding to integrins. 47 Moreover, collagens and laminins are a source of bioactive fragments, matrikines, that are released due to the activity of proteinases during ECM remodeling.48,49 We identified nephronectin as the most abundant glycoprotein in the lung ECM scaffold. Of note, nephronectin has been observed in the lung during development 26 and is particularly enriched in IPF [32]. This abundant and widespread expression of this ECM glycoprotein in the lung suggests a bioactive role that requires further investigation. The use of mass spectrometry to characterize ECM scaffolds provides a high-throughput analysis of ECM composition by the identification of the full proteomic repertoire preserved after decellularization. Using this approach, we are able to obtain a better resolution of ECM composition and, consequently, to identify new and less abundant lung ECM components, such as the MEGF6.
ECM proteins, such as collagen and fibronectin, are commonly used alone or in combination to facilitate cell adhesion and growth of primary lung cells in vitro.32,50 Decellularized ECM tissue coatings from brain, 51 cardiac, and skeletal muscle 52 were described in literature. In these studies, pepsin was used for ECM enzymatic dissociation. However, this broad-spectrum enzyme is not naturally present in the lung, and the trypsin-generated ECM peptides do not represent the in vivo ECM degradation products formed in the context of the lung. As an alternative, here we propose the use of mechanical ECM homogenization. When this ECM preparation was applied as a protein coating on TCPS, in standard 2D culture conditions, lung ECM supported attachment of LFs. These findings advocate the potential use of homogenized lung ECM for surface modification, to promote attachment, and to increase cytocompatibility of synthetic scaffolds. To assess the biochemical and structural impact of the generated ECM on primary LFs, we established a 3D-like culture that allows live cell imaging and confocal analysis. LFs attached to the ECM, formed cell protrusions in close contact to the ECM, proliferated, and repopulated the decellularized lung scaffold. Electron microscopic images show that cells attached to delicate topological structures on the surface of ECM scaffolds.
Diverse mechanical and biochemical signals, such as ECM stiffness, are mediated by the transcriptional coactivators YAP/TAZ signaling, which strongly impact cell fate. 53 YAP was distributed through the cytoplasm and nuclei in the 2D setting; whereas in the lung, ECM YAP was translocated to the nuclei, indicating activation of YAP-mediated signaling. However, cells are not regulated by the ECM in a passive manner; in fact, the reciprocal interaction between cells and the surrounding ECM is of utmost importance. Cells sense the microenvironment and remodel the ECM by secreting ECM structural proteins, such as collagens, while also degrading the matrix by MMPs. 6 We show that LFs produce an activated form of MMP-2 and MMP-9 when cultured in vitro on lung ECM scaffolds. Activated MMP-2 is reported to directly degrade collagen I, 54 whereas a variety of ECM proteins including fibronectin, laminin, and collagen are proteolytically processed by MMP-9 55 as part of the ECM remodeling process.
The herein developed 3D-like slice culture system mimics the composition and organization of the in vivo extracellular microenvironment. This system can be readily prepared and requires only standard histological equipment. Isolated ECM explants do not require snap freezing or washing in concentrated ethanol before use as culture substrates,15,34 the processes that may negatively affect the structure and biochemical properties of the ECM. Due to their relatively small thickness (25 μm), the slices do not require further sectioning for immunostaining and microscopic analysis, unlike in thicker slices,34,35 and can be immunostained and visualized under a confocal microscope directly in an Ibidi culture dish. The use of thick slices ensures that all anatomical structures of lung ECM are available for cell–matrix interaction and that cells become exposed to ECM components organized at nano and microscale. However, 25 μm slices are too thin to provide a real 3D environment for the cells as they do not become completely embedded in the ECM, in contrast to native tissue in which LFs reside in nonpolarized environments. Hence, this 3D-like lung decellularized culture system constitutes a simple and practical tool to dissect the interaction of lung cells with the extracellular microenvironment in close-to-native conditions. This system can be easily applied to other organ systems and disease states. Indeed, we have recently reported a similar protocol for heart 19 and intestine 56 decellularization in mouse and human samples, in normalcy 19 and disease. 56 This article constitutes a proof-of-principle study highlighting the relevance of 3D-like culture on decellularized lung ECM for the understanding of the reciprocal interaction between lung ECM and LFs in healthy vs disease states. In addition, these approaches can be also used as drug screening platforms.
Conclusions
In this study, we developed a 3D-like in vitro culture platform to study cell–ECM biological interactions in a native-like environment. Our decellularization protocol, which does not require complex perfusion equipment, generates biocompatible and bioactive acellular lung scaffolds with preserved ECM architecture and composition. Proteomic analysis of decellularized lung scaffolds identified a plethora of ECM and ECM-associated molecules conserved in ECM scaffolds. A practical system using thin slices of decellularized ECM was established for seeding of LFs that was shown, in a proof-of-principle experiment, to be suitable for imagining cell–ECM interactions at the morphological and molecular level. The herein described 3D-like model will be particularly valuable for dissecting pathological and physiological mechanisms of lung cell–ECM crosstalk.
Footnotes
Acknowledgments
The authors acknowledge the Animal House caretakers from i3S and Masaryk University. They also acknowledge the core facility cellular Imaging CELLIM of CEITEC—Central European Institute of Technology supported by the Czech-Bioimaging large RI project (LM2015062 funded by MEYS CR) for their support with obtaining scientific data presented in this article. This work was supported by INFARMED—Autoridade Nacional do Medicamento e Produtos de Saúde, I.P. [FIS-FIS-2015-01_CCV_20150630-157]; by Norte Portugal Regional Operational Programme (NORTE 2020), under the PORTUGAL 2020 Partnership Agreement, through the European Regional Development Fund (ERDF) [NORTE-01-0145-FEDER-000012]; by European Structural and Investment Funds (ESIF), under Lisbon Portugal Regional Operational Programme and National Funds through FCT—Foundation for Science and Technology [POCI-01-0145-FEDER-016385]; by FCT—Fundação para a Ciência e a Tecnologia/Ministério da Ciência, Tecnologia e Inovação in the framework of the project “Institute for Research and Innovation in Health Sciences” [POCI-01-0145- FEDER-007274]; by the Ministry of Health of the Czech Republic [grant no. 16-31501A]; by the National Program of Sustainability II [LQ1605, MEYES CR]; by funds from the Faculty of Medicine MU to junior researcher (Z.K.); by the project TissueEng7 of Faculty of Medicine MU [MUNI/A1369/2016]; and by an individual fellowship to [SFRH/BD/88780/2012] to A.C.S. CIISB research infrastructure project LM2015043 funded by MEYS CR is gratefully acknowledged for the financial support of the mass spectrometry measurements at the Proteomics Core Facility. D.P. and Z.Z. are thanked for their financial support to the Czech Science Foundation [P206/12/G151]. The work of V.V. was supported by the European Social Fund and European Regional Development Fund—Project MAGNET (No. CZ.02.1.01/0.0/0.0/15_003/0000492).
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.