
Abstract
In vitro
three-dimensional (3D) cultures of hepatocytes are increasingly being used to assess human hepatic metabolism and toxicity in drug development. In this study, we developed an in vitro 3D cell culture method with a microstructured mesh sheet and applied it to culturing human hepatoma HepG2 cells. The micromesh sheet is constituted of fine mesh strands and apertures that are each much larger than a single cell in size. Proliferating on a micromesh sheet, HepG2 cells spread out in a planar manner and then formed a multilayered cell sheet, so that cell–cell adhesion was dominant over cell–substrate adhesion as being different from two-dimensional (2D) cultures. In micromesh cultures, the increase rate in thickness of the cell mass was visually slower than that in spheroid cultures, enabling us to clearly observe inside cells of the cell population by microscopy. Micromesh-cultured HepG2 cells showed higher viability compared with spheroid-cultured cells. The multilayered HepG2 cell sheet increased expression of hepatic marker genes and induced cell polarization with bile canalicular membranes. Furthermore, a combination of micromesh cultures with medium perfusion further induced expression of hepatic marker genes in HepG2 cells; especially cytochrome P450 1A1 (CYP1A1) and CYP1A2 messenger RNA (mRNA) increased 86-fold and 43-fold compared with 2D controls, respectively, which were much higher than those in spheroid cultures. Thus, this simple and versatile micromesh culture method holds some advantages over traditional spheroid cultures and is expected to be instrumental in culturing more differentiated hepatic cells such as HepaRG cells and primary hepatocytes for future preclinical testing.
Impact Statement
Spheroid cell cultures are extensively used in three-dimensional (3D) culture methods, but have some technical difficulties in the microscopic cell observation, the organization of cell layer structures, and the supply of oxygen and nutrients in the core of spheroids. This study presents novel 3D cultures of which cells are simply grown on a micromesh sheet. Micromesh-cultured cells form thin sheet-like layers with improvement in the abovementioned difficulties of spheroid cultures and promise to be methods for modeling tissues in future multiple cell-based assays.
Introduction
Drug development requires information on human hepatic metabolism to accurately predict drug toxicity and efficacy in humans. 1 Animal models are useful tools to assess hepatic metabolism of chemicals in vivo but their prediction accuracy for human metabolism is limited, and animal experiments naturally accompany ethical issues. In vitro cell culture models with human hepatocytes or human hepatoma cell lines such as HepG2 cells have been developed to complement or replace animal models. Traditional in vitro two-dimensional (2D) cell cultures have been used for a long time in drug development, but 2D cultures induce mechanical stimuli to cells, which affect tissue-specific phenotypes.2,3 For example, HepG2 cells express drug-metabolizing enzymes at low levels in 2D cultures, 4 and primary hepatocytes rapidly lose the metabolic activity after beginning 2D cultures.5,6
In vitro three-dimensional (3D) cell cultures are recognized as more structurally and functionally authentic cultures, which enable cells to partially retain hepatic phenotypes. 7 Spheroid cultures are well-established 3D cell cultures, and many methods to create spheroidal cells have been developed so far, for example, spheroid cultures with hanging drop plates, 7 round bottom plates, 8 Matrigel, 9 or agarose microwell plates. 10 However, spheroid cultures have disadvantages that come from their thickness. As HepG2 spheroids exceed around 200 μm in diameter, the oxygen supply becomes insufficient in the core of spheroids, leading to hypoxia-induced necrosis,11–13 and it also becomes harder to observe inside cells by microscopy. Moreover, the structure of spheroids is not suitable to recapitulate layer structures of tissues in vivo.
A cell culture technique with a microstructured mesh sheet, named micromesh culture, was originally developed with our notion that minimization of cell-basal substrate adhesion can promote cellular functions. 14 The micromesh sheet is constituted of mesh apertures that are each larger than a single cell. Cells can form a 2D cell sheet following cell inoculation and proliferation on the micromesh sheet. Our previous studies yielded interesting results that human induced pluripotent stem (iPS) cells can spontaneously differentiate into trophoblast lineage cells on a micromesh sheet without stimulation with cytokines or small molecules.14,15 In the present study, we tried to expand this micromesh culture technique to make a 3D cell sheet and analyze it. We hypothesized that micromesh cultures enable cells to form a 3D cell sheet, which should be advantageous over 3D spheroids in terms of oxygen and nutrient supply and cell observation, and to promote differentiation or maturation in other kinds of cells, including hepatic cells.
In this study, we applied micromesh cultures to human hepatoma HepG2 cells. On a micromesh sheet, HepG2 cells formed a multilayered cell sheet of which cells were analyzed by both bright-field and fluorescence microscopy more clearly than spheroidal cells. Cell viability was higher in micromesh cultures than in spheroid cultures. Micromesh cultures induced polarization of HepG2 cells, which were determined by localization of multidrug resistance-associated protein 2 (MRP2) proteins and accumulation of fluorescein-labeled bile acids. Expression of some hepatic marker genes such as cytochrome P450 1A1 (CYP1A1) gene and CYP1A2 gene was significantly induced by micromesh cultures more than spheroid cultures in HepG2 cells. Furthermore, a combination of the micromesh cultures with perfusion cultures further promoted such induction. We propose that these simple and versatile micromesh culture techniques have great potential for cell observation and differentiation in future preclinical studies.
Materials and Methods
Fabrication of micromesh sheets
Microstructured mesh sheets were fabricated by standard photolithography with nickel (Optnics Precision Co., Ltd., Tochigi, Japan) and coated with Parylene (DPXC, CAS No. 28804-46-8; Parylene Japan, Tokyo, Japan) using a PDS-2010 Labcoter 2 (Specialty Coating Systems, Inc., Indianapolis, IN). Parylene, a biocompatible material, was used to avoid a nickel leak from the mesh sheet. The micromesh sheet was constituted of triangle- or rhombus apertures (50–200 μm in pitch) and fine mesh strands (5 μm in width).
Micromesh sheets of polydimethylsiloxane (PDMS; Silpot184; Toray-Dow Corning, Tokyo, Japan) were fabricated with a nickel micromesh sheet as a mold. PDMS elastomer and curing agent were mixed in the weight ratio of 10:1, and the mixture was diluted 13-fold with hexane. A nickel micromesh sheet was spin-coated with the diluted PDMS solution and baked. This coating/baking process was performed twice. Then, nickel was etched with 0.1 M HCl. Finally, the PDMS micromesh sheet without nickel was washed and dried.
PDMS fluidic devices
PDMS fluidic devices were fabricated by soft lithography. A 3D printer (AGILISTA-3200; Keyence, Osaka, Japan) was used to make molds for PDMS fluidic devices. PDMS (elastomer: curing agent = 10:1) was poured into the molds and baked to solidify them. After taking off the solidified objects from the molds, silicone tubes (outer diameter 2 mm × inner diameter 1 mm) were connected to the PDMS fluidic devices. Medium perfusion was performed using a syringe pump (KD Scientific, Holliston, MA).
Procedure of cell seeding on a micromesh sheet
A silicon rubber sheet was punched to make a hole (ϕ 4 mm), and the hole was covered by a micromesh sheet. The micromesh sheet was fixed using a fixing Kapton tape of which thickness is <100 μm (P-222 AMB; Nitto, Osaka, Japan). To sandwich the micromesh sheet with two silicon sheets, an additional silicon sheet with the same hole was put on the mesh sheet. As a drop of cellular suspension, cells were seeded on the micromesh sheet from above, and surface tension retained cells on the micromesh sheet. After 4 h, the combined construct was transferred, leaving the bottom silicon sheet, onto two spacers in a well. Then the well was filled with culture media.
Cell line and culture conditions
Human hepatoma cell line HepG2 cells (No. RCB1886) were purchased from RIKEN BRC (Tsukuba, Japan). HepG2 cells were cultured at 37°C with 5% CO2 in Dulbecco's modified Eagle's medium (Thermo Fisher Scientific, Waltham, MA) supplemented with 10% fetal bovine serum (Gibco), 100 U/mL penicillin, and 100 μg/mL streptomycin. HH cells, bovine carotid artery normal endothelial cells (No. JCRB0099), were purchased from Japan Collection of Research Bioresources (JCRB) Cell Bank (Osaka, Japan) and cultured under the same condition as HepG2 cells.
Cell seeding and cultures
For micromesh cultures, cells were seeded on a micromesh sheet at 1 × 105 cells/ϕ 4 mm circle in wells of a 12-well plate. Cells were cultured for 5 days, and culture media (2 mL/well) were changed every other day. For fluidic micromesh cultures, PDMS fluidic devices shown in Figure 6 and Supplementary Figure S2 (Supplementary Data are available online at
Cocultures with HepG2 cells and HH cells
First, endothelial HH cells were stained with 1.25 μg/mL DiI (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarboxyamine perchlorate; Setareh Biotech, Eugene, OR) and seeded onto a PDMS micromesh sheet (ϕ 4 mm circle). HepG2 cells stained with a CellBrite Green Cytoplasmic Membrane-Labeling Kit (80-fold dilution; Biotium, Inc., Hayward, CA) were seeded on HH cells 24 h later. Cells were washed with phosphate buffered saline (PBS) (-) 24 h later and then fixed with 4% paraformaldehyde (PFA) for 20 min. Cells were washed, mounted with VECTASHIELD solution containing 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories, Burlingame, CA), and coverslipped. The cell sheet consisting of HepG2 cells and HH cells was analyzed using a Zeiss LSM 800 confocal microscope (Carl Zeiss, Oberkochen, Germany).
Cell staining
HepG2 cells cultured in spheroid cultures or micromesh cultures for 5 days were stained with hematoxylin and eosin (HE). As 2D controls, cells were cultured on cover glasses and stained. Spheroids on agarose gel plates were transferred onto cover glasses 1 day before HE staining to attach spheroids on them. On day 5 after cell seeding, cells were washed with PBS(-) and Milli-Q water, followed by staining with Mayer's hematoxylin solution (Wako, Osaka, Japan) for 5 min. Cells were washed with 0.08–0.09% ammonia solution (Wako) for 3 min and then washed by water (ca. 50°C) for 7 min. Cells were stained with 1% Eosin Y solution (Wako) for 3 min and washed with ethanol several times (95% thrice, 100% thrice). Then, cells were treated with xylene for 3 min and mounted with Histomount (National Diagnostics, Atlanta, GA). HE-stained cells were analyzed using a BZ-X710 All-in-one fluorescence microscope (Keyence).
For staining cells with DiI, micromesh-cultured HepG2 cells were incubated in culture media containing 0.05 mg/mL DiI for 20 min on day 5 after seeding. Cells were washed by PBS(-) and then fixed with 4% PFA for 20 min. Cells were washed once, mounted with VECTASHIELD/DAPI, coverslipped, and analyzed by confocal microscopy.
Cell viability assay
HepG2 cells were cultured in spheroid cultures or micromesh cultures for 5 or 8 days. As 2D controls, cells were seeded on cover glasses in wells. Culture media (2 mL/well) were changed every other day. To count live or dead cells in cell populations, cells were washed with PBS(-), treated with trypsin for 15 min, and then suspended in media. The media were removed after centrifugation, and the cellular pellet, including live or dead cells, was resuspended in 100–200 μL of media. Dead cells were stained with 0.2% trypan blue. Stained or nonstained cells were counted using Countess II Automated Cell Counter (Thermo Fisher Scientific).
Immunocytochemistry
HepG2 cells were washed with PBS(-) and fixed in 4% PFA solution for 20 min. For permeabilization, cells were treated with PBS(-) containing 0.1% Triton X-100 for 30 min. Cells were washed, blocked with 5% bovine serum albumin in PBS(-) containing 0.1% Tween 20 (PBS-T) for 20 min, and incubated with a given primary antibody overnight. Primary antibodies against MRP2 (ab3373, 1:50 dilution), albumin (ALB; ab10241,1:500), and cytochrome P450 1A (CYP1A; ab22717, 1:500) were purchased from Abcam (Cambridge, MA) and used in this study. Cell samples were incubated with a secondary antibody (Alexa 488 anti-mouse IgG Fab2, 1:500 dilution; Cell Signaling Technology, Danvers, MA), washed with PBS-T, mounted with VECTASHIELD/DAPI, coverslipped, and imaged by confocal microscopy.
Analysis of bile acid transport and MRP2 localization
HepG2 cells were seeded on a micromesh sheet at 1 × 104 cells/ϕ 4 mm circle in wells of a 12-well plate. Cells were cultured for 5 days, and culture media (2 mL/well) were changed every other day. As a 2D control, cells were cultured on a cover glass. HepG2 spheroids on an agarose gel plate were transferred onto a cover glass 1 day before analysis of bile acid transport. On day 5, cells in each culture were washed with Hanks' balanced salt solution (HBSS; Thermo Fisher Scientific) and incubated with 1 μM cholyl-lysyl-fluorescein (CLF), a fluorescein-labeled bile acid (Corning Life Sciences, Tewksbury, MA), for 30 min. Cells were washed with HBSS thrice, incubated in HBSS with 1 μg/mL Hoechst 33342 (Thermo Fisher Scientific), and imaged by confocal microscopy. To analyze MRP2 localization, cells were cultured under the same conditions as the bile acid transport analysis. Cells were washed with PBS(-) on day 5, followed by immunocytochemistry described above.
Real-time polymerase chain reaction
HepG2 cells were cultured for 5 days; for cell images, see Supplementary Figure S3B. Total RNA was extracted from HepG2 cells using an RNeasy Micro Kit (Qiagen, Valencia, CA). Complementary DNA (cDNA) was synthesized with the RNA and a ReverTra Ace qPCR RT Kit (Toyobo, Osaka, Japan). Real-time polymerase chain reaction (PCR) was conducted using a Power SYBR Green Master Mix (Applied Biosystems, Foster, CA), gene specific primers (Supplementary Table S1), and QuantStudio 5 Real-Time PCR System (Applied Biosystems). Messenger RNA (mRNA) expression levels were normalized with β-actin mRNA expression levels.
Enzyme-linked immunosorbent assay for albumin measurement
Albumin secretion levels from HepG2 cells were measured using a human albumin enzyme-linked immunosorbent assay (ELISA) kit (Bethyl Laboratories, Inc., Montgomery, TX). HepG2 cells were cultured in wells of a 12-well plate, on agarose plates with 256 wells, or on micromesh sheets, for 5 days, and culture media were changed every other day. On day 5, culture media in which cells were cultured for 18 h were collected to measure the amount of albumin secreted. For fluidic micromesh cultures, cells were seeded on micromesh sheets, and each mesh sheet was placed on a fluidic device shown in Figure 6. Culture media constantly flowed at 1 mL/day in the fluidic device. Micromesh sheets in devices were transferred into wells of a 12-well plate 18 h before collecting culture media. Results of ELISA were normalized by the number of cells that were estimated by measuring protein concentrations with a Pierce BCA Protein Assay Kit (Thermo Fisher Scientific).
Statistical analyses
Multiple groups and two groups were analyzed by Tukey's multiple comparison test and Student's t-test, respectively. A value of p < 0.05 was considered statistically significant. These statistical analyses were conducted using StatView version 5.0.1 (SAS Institute, Inc., Cary, NC).
Results
Micromesh cultures of HepG2 cells
First, to seed cells, a drop of HepG2 cell suspension was placed on a variety of micromesh sheets, 50–200 μm in pitch (Fig. 1A–D). Cells were attached to fine mesh strands in all types of micromesh sheets (Fig. 1E and Supplementary Fig. S1). Unexpectedly, as observed in mesh sheets with larger apertures (100–200 μm in pitch), surface tension of the drop and strong adhesiveness of HepG2 cells helped cells to be placed within mesh apertures. Cells then proliferated and filled the opening spaces of the mesh sheets, resulting in forming cell sheets (Fig. 1E and Supplementary Fig. S1). Larger apertures were suitable for the observation of cell shapes, and smaller apertures were available to form even HepG2 cell sheets at the early stage after cell seeding.

In vitro cell culture method with a micromesh sheet.
Cell analysis with transmitted light provides important information, including intra- or extracellular structures and intercellular junctions. However, 3D spheroids have generally technical difficulties in analysis with transmitted light due to their thickness and therefore are analyzed as sections following paraffin embedding. Figure 2A shows HepG2 cells stained with HE that is widely used in histology. Cell nuclei of micromesh-cultured cells or 2D monolayer cells were clearly observed, whereas nuclei of spheroidal cells were unclear (Fig. 2A). The microscopic analysis of HE-stained cells indicated that HepG2 cells cultured for 5 days by micromesh cultures formed a multilayered cell sheet as shown in Figure 2B. To image the multilayered cell sheet accurately, cell nuclei and membrane were stained with DAPI and DiI, respectively, followed by fluorescence confocal microscopy. HepG2 cells cultured for 5 days were found to form a cell sheet of 60–70 μm thickness (Fig. 2C). Taken together, micromesh-cultured 3D cells can be visually analyzed easier than 3D spheroids.

Differences in microscopy or cell viability between 2D-, spheroid-, and micromesh-cultured cells.
Differences in cell viability between 2D-, spheroid, and micromesh-cultured cells
We examined whether micromesh cultures improved the supply of oxygen and nutrients to cells against spheroid cultures. HepG2 cells were cultured in 2D spheroid cultures or micromesh cultures and were subjected to cell viability assay. Results indicated that cells in each culture method did not die significantly when they were cultured for 5 days with changing media every other day (Fig. 2D). Differences in cell viability were observed when cells were cultured for 8 days with medium changes. The Live/Dead ratios of 95%/5% and 92.5%/7.5% were determined for 2D cultures and micromesh cultures, respectively. However, spheroid cultures showed significantly lower values (84%/16%). Cells in the core of spheroids were darker than cells in micromesh cultures (Supplementary Fig. S3A). Together with previous findings that hypoxia-induced necrosis can occur in the core of spheroids, these results suggested that micromesh cultures improve the oxygen supply to cells.
Formation of a multilayered cell sheet
The hepatic lobule in vivo is constituted of parenchymal and nonparenchymal hepatocytes such as endothelial cells and locally organizes layered structures consisting of them. 6 When cells are layered by usual methods in vitro, cells forming the bottom layer are attached to a basal substrate, provoking a high mechanical stress to the basal cells. To organize different kinds of cells as a multilayered cell sheet like tissues in vivo, that is, cell sheets without strong cell-basal substrate adhesion, normal endothelial HH cells and HepG2 cells were cocultured with a PDMS micromesh sheet. HH cells and HepG2 cells were stained with DiI and a green fluorescent dye, respectively. Since micromesh sheets coated with Parylene have strong autofluorescence, they are unsuitable in this fluorescence microscopy. Contrary to Parylene, PDMS hardly has autofluorescence, and therefore, PDMS micromesh sheets were developed (Fig. 3A). First, HH cells were seeded on a PDMS micromesh sheet, and HepG2 cells were seeded on HH cells 24 h later (Fig. 3B). A multilayered cell sheet was clearly imaged by confocal microscopy (Fig. 3C), which seemed to partially recapitulate a hepatocyte-endothelial cell layer in the hepatic lobule. This layer system may be instrumental in studies of hepatic events stimulated by direct interactions with nonparenchymal hepatocytes.
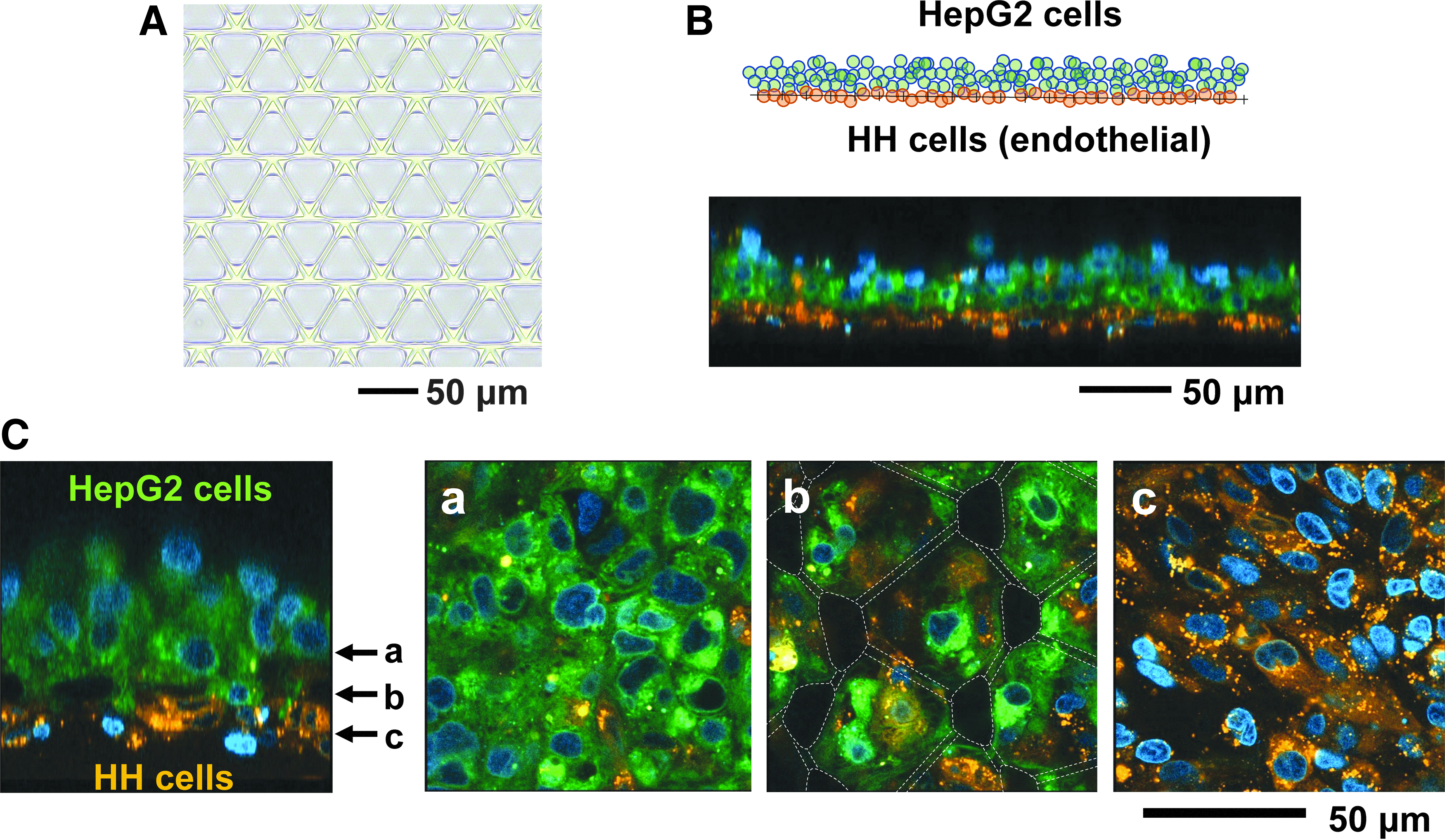
A multilayered cell sheet consisting of HepG2 cells and HH cells.
Structural changes of 3D spheroids on a micromesh sheet
HepG2 spheroids stained with a green fluorescent dye were placed on a PDMS micromesh sheet to examine their structural changes. On the mesh sheet, the shape of spheroids remained almost unchanged during a few days, while spheroids attached and changed their shape on a 2D basal plate sooner (Fig. 4A, B). Spheroids on the micromesh sheet appeared to be a bowl as observed by confocal microscopy since the laser beam could not reach the depths and the upper parts of the spheres (Fig. 4B). Spheroids changed into planar shapes 5 days after starting culture on the mesh sheet, and the structure of the cell sheet was able to be imaged in whole.
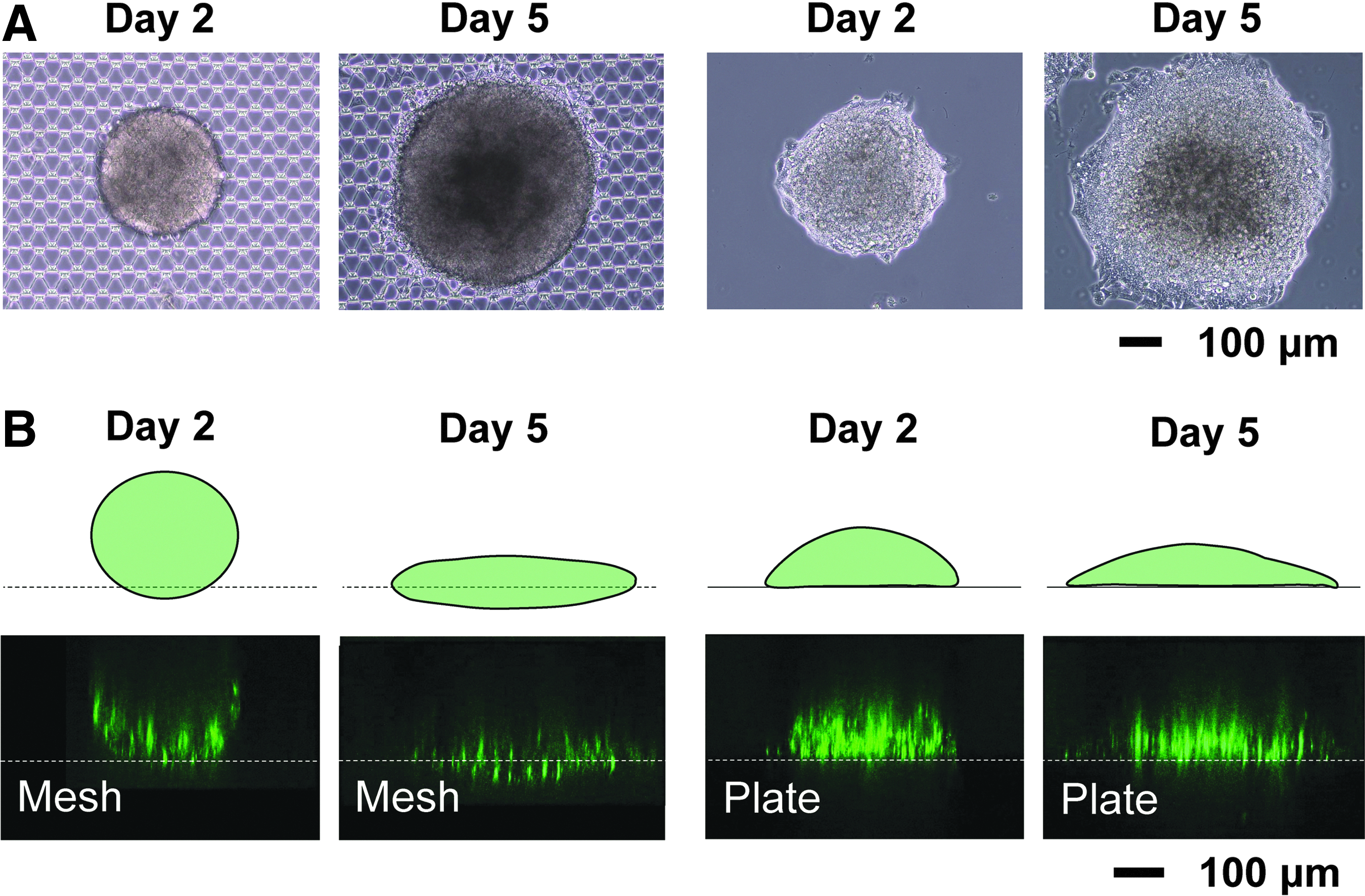
Structural changes of 3D spheroids on a micromesh sheet.
Localization of MRP2 and accumulation of bile acids in micromesh-cultured HepG2 cells
Hepatocytes have the apical–basolateral polarity, and the apical membranes form bile canaliculi between the cells. Hepatocytes secrete bile, including bile acids, into bile canaliculi through MRP2 that is mainly localized in the apical membranes. HepG2 cells in 2D cultures cannot exhibit hepatocyte polarity, but HepG2 spheroids are known to restore the polarity in part. 9 To analyze MRP2 localization fluoroscopically, HepG2 cells were cultured on cover glasses, on agarose gel microwell plates to make spheroids, or on PDMS micromesh sheets and then stained with an anti-MRP2 antibody. MRP2 was localized on cell membranes among cells in spheroid cultures and micromesh cultures, but not in 2D cultures (Fig. 5A). Some localized MRP2 formed distorted spheres. A fluorescein-labeled bile acid CLF, which can be transported by MRP2 to bile canaliculi, was accumulated in spheroid-cultured or micromesh-cultured HepG2 cells, but not 2D-cultured cells (Fig. 5B). Some cell nuclei are out of focus in Figure 5B, Mesh. The CLF accumulation pattern appeared to resemble the MRP2 localization pattern. Thus, these results demonstrated that HepG2 cells partially regain its polarity by micromesh cultures, as well as by spheroid cultures.

Localization of MRP2 proteins and accumulation of bile acids.
Enhancement of hepatic functions by static- or fluidic-micromesh cultures
We assessed impacts of micromesh cultures on functions of HepG2 cells based on expression levels of marker genes for mature hepatocytes. We prepared micromesh-cultured HepG2 cells not only in wells but also in fluidic devices (Fig. 6), since medium perfusion is known to increase functions of hepatic cells.
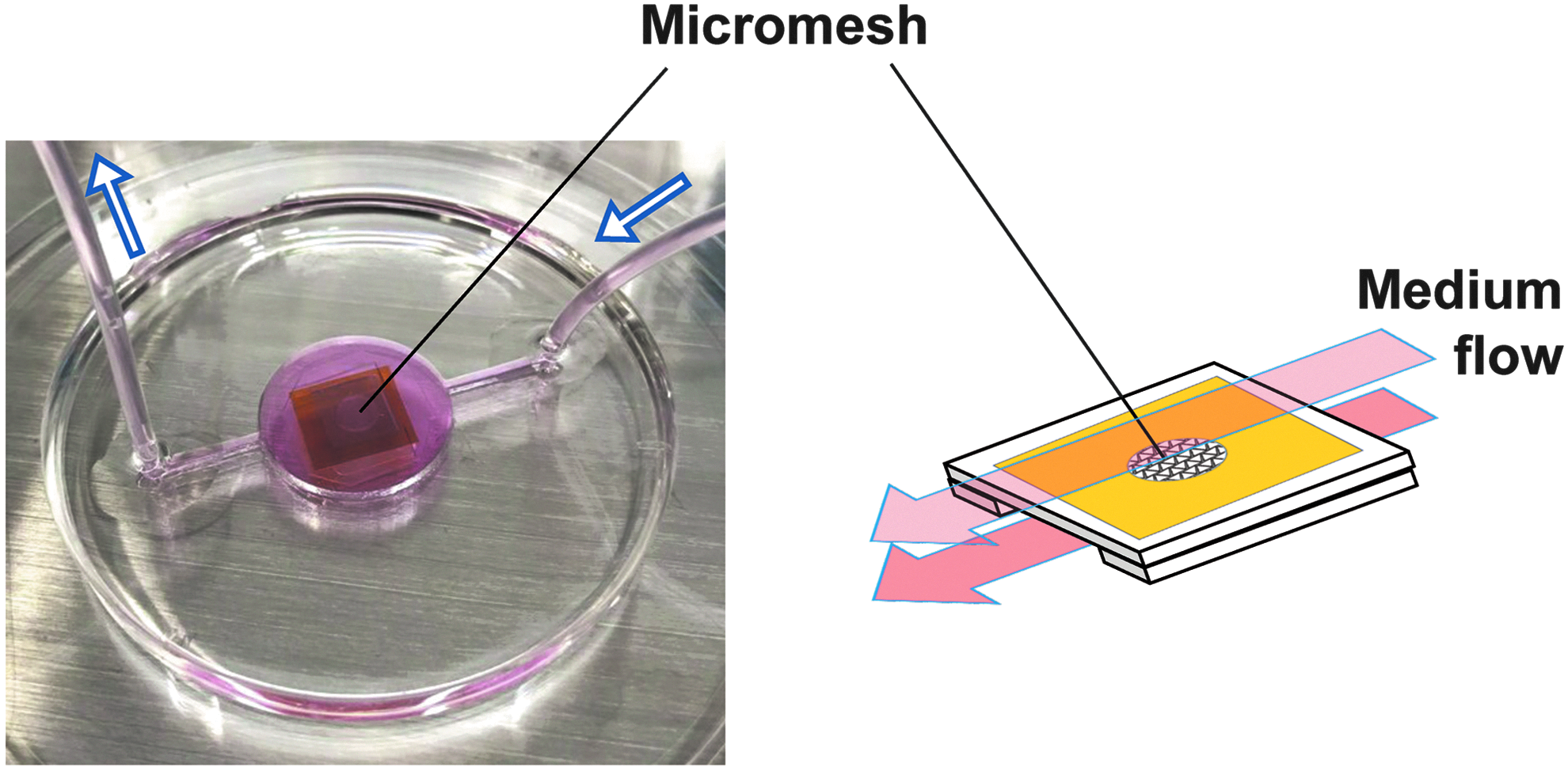
A PDMS microfluidic device for fluidic micromesh cultures. A PDMS device was fabricated for fluidic micromesh cultures. A micromesh sheet supported by two spacers was placed in the center of the device, and fresh media were perfused from one side to the other (from right to left in this figure) using a syringe pump. Fresh media pass through the upper and lower sides of the cell/micromesh sheet (shown as arrows in red).
In static conditions, micromesh cultures induced expression of ALB mRNA and α1-antitrypsin (α1-AT) mRNA as spheroid cultures did (Fig. 7A). mRNA expression levels of CYP2B6, UGT1A1, and UGT2B4 were slightly higher in static micromesh cultures than in spheroid cultures, but those of CYP2C9, CYP3A4, and MRP2 were not (Fig. 7A). Interestingly, CYP1A1 mRNA and CYP1A2 mRNA were markedly induced by static micromesh cultures compared with spheroid cultures. mRNA expression levels of aryl hydrocarbon receptor (AhR), constitutive active/androstane receptor (CAR), and pregnane X receptor (PXR), which regulate transcription of drug-metabolizing enzyme genes, did not change significantly in these cultures (Supplementary Fig. S4A). Parylene coating to avoid a nickel leak from micromesh sheets and energetic plasma treatment to increase hydrophilicity were confirmed not to affect expressions of ALB and CYP1A2 genes (Supplementary Fig. S4B). Aperture sizes of micromesh sheets also did not make a difference in these expressions in this study (Supplementary Fig. S4C). At protein levels, ALB and CYP1A were detected on micromesh sheets (Supplementary Fig. S5), and enhanced ALB secretion was confirmed in spheroid or micromesh cultures (Fig. 7B).
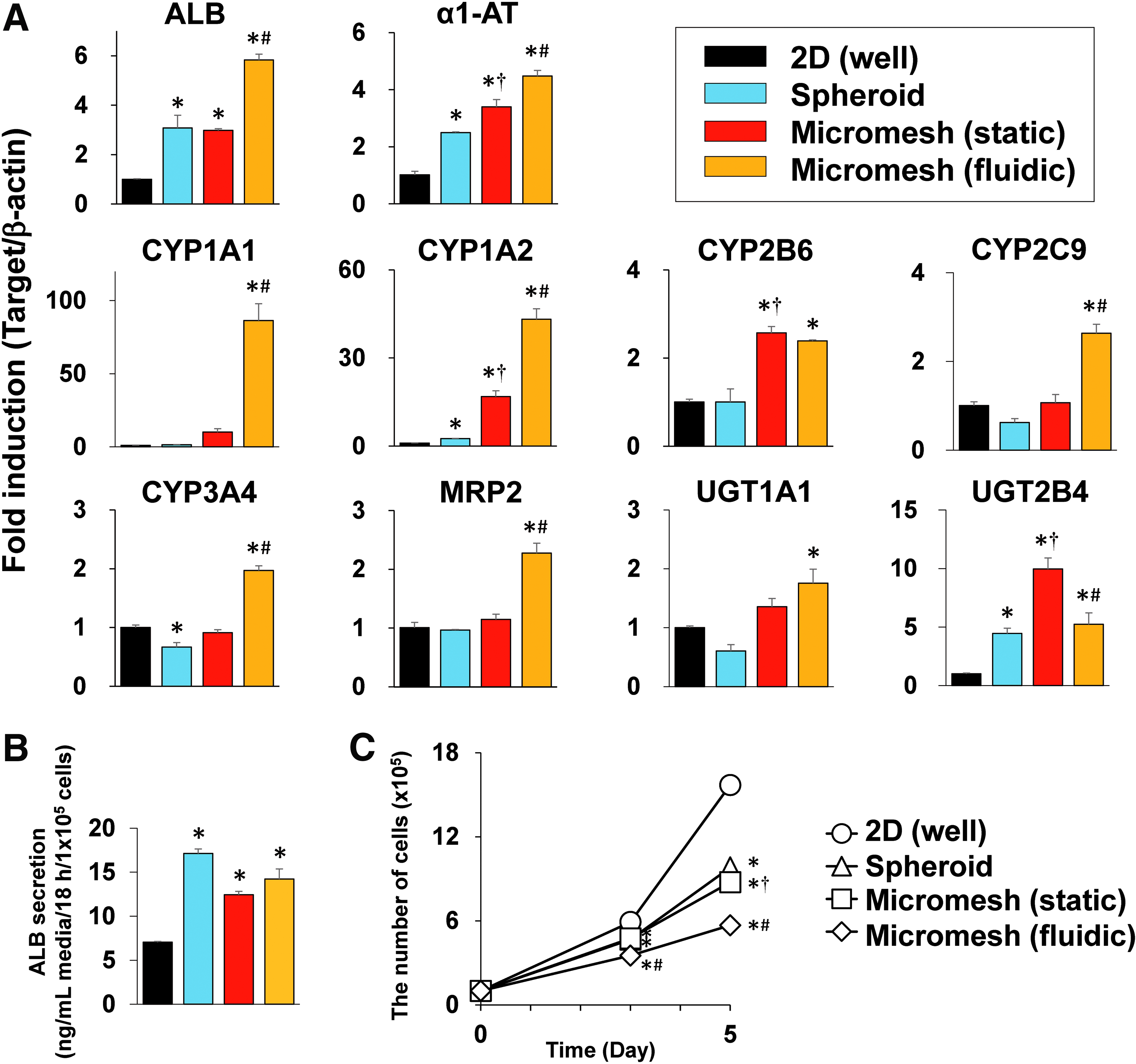
Functional regulation in HepG2 cells by micromesh cultures.
In perfusion cultures, HepG2 cells were cultured on the micromesh sheet with a continuous medium flow at 1 mL/day for 5 days. The fluidic micromesh cultures further promoted expressions of hepatic marker genes compared with static micromesh cultures (Fig. 7A; yellow bars). A fluidic device of different shape, which was made to easily transfer a micromesh sheet into or out of the device at any time, showed the almost same results as the chip-type device showed (Supplementary Fig. S4D).
The proliferation rate of HepG2 cells was assessed, since proliferation of differentiated HepG2 cells is known to cease. 9 Proliferation of micromesh-cultured or spheroid-cultured HepG2 cells was significantly delayed (Fig. 7C). Fluidic micromesh cultures were more effective in delaying it. Collectively, these results indicate that static or fluidic micromesh cultures enhance hepatic functions of HepG2 cells.
Discussion
In vitro 3D cell culture models are becoming increasingly important in drug development under the current circumstances with 3Rs strategy (replacement, reduction, and refinement) on animal experiments. In this study, we have developed a 3D cell culture named micromesh culture. As features of micromesh cultures, micromesh-cultured HepG2 cells partially expressed considerably differentiated phenotypes than spheroid-cultured HepG2 cells, implying that micromesh cultures regulate cellular mechanotransduction. Micromesh-cultured cells showed a multilayered cell structure that began from a monolayer and did not show a spherical shape like a spheroid. Therefore, cells inside the cell mass were clearly observed by microscopy. Since, in micromesh cultures, cells after division spread out in a planar manner along the micromesh sheet, the increased rate in thickness of the cell mass was visually slower than that in spheroid cultures, which resulted in higher cell viability on the mesh sheet. These findings suggest that micromesh cultures hold advantages over traditional spheroid cultures.
In HepG2 cells, expression levels of the CYP1A1 and CYP1A2 genes were much lower than those in human primary hepatocytes,16,17 and strong increases in those expressions by micromesh cultures show signs of differentiation in HepG2 cells. The molecular mechanisms of the increases are not revealed yet. AhR is a representative transcriptional factor regulating expression of CYP1A1/2 genes, 18 but its expression was not changed by micromesh cultures (Supplementary Fig. S4A). Given that AhR generally exists in an inactive state in the absence of ligand binding, the increases in CYP1A1/2 should not be elicited by AhR. Spheroid cultures increased CYP1A1/2 mRNA, but the increased levels were much less than those by micromesh cultures, suggesting that the striking increases by micromesh cultures arose from a factor other than the enhanced cell–cell interactions. Since micromesh-cultured HepG2 cells form a cell sheet that is thinner than 3D spheroids, the efficient supply of oxygen and nutrients may contribute to the marked CYP1A1/2 mRNA induction. This hypothesis seems to be supported by the finding that fluidic micromesh cultures further enhanced the induction.
Micromesh cultures can be applicable to organs-on-chips, which are microfluidic cell culture systems recapitulating physiological functions and structures of living organs. Organs-on-chips are expected to be powerful tools to predict drug toxicity and efficacy in preclinical studies and to serve to understand microphysiological phenomena which could not be studied with existing 2D cell culture models. 19 For instance, lung-on-a-chip was able to recapitulate physiological behaviors of neutrophils and to reproduce drug toxicity-induced pulmonary edema observed in cancer patients treated with interleukin-2.20,21 It should be noted, however, that the PDMS membrane within the lung-on-a-chip is basically low-porosity (it appears <25%), and therefore, cells attaching on the membrane should be cultured under high mechanical stimuli. Our PDMS micromesh sheet with high porosity can be substituted for their low-porosity membrane to reduce mechanical stimuli from cell–substrate adhesion.
In hepatocytes, 3D spheroid cultures and perfusion cultures can each sustain tissue specific functions at high levels for long periods of time, and integration of 3D-cultured cells into fluidic devices synergistically works to promote cellular functions.22–24 When 3D spheroids are incorporated into a fluidic device, spheroids are required to be anchored within the device to stand in shear flow without changing their structure from 3D to 2D. To tether them, researchers often use customized bioinks containing, for example, extracellular matrix. 25 However, such a culture matrix may disturb downstream experimentation in some cases. A micromesh sheet was able to anchor HepG2 spheroids as in 3D cells without any bioinks, while spheroids gradually changed into 2D structures on a basal plate (Fig. 4A, B). Thus, a micromesh sheet is available as an anchor for spheroids within fluidic devices. Taken together with the findings that micromesh-cultured cell sheets can be easily put into fluidic devices, micromesh sheets are structurally suitable for 3D cell cultures in fluidic devices. Micromesh cultures in such fluidic devices with more differentiated HepaRG cells, iPS-derived hepatocytes, or primary hepatocytes are important experiments in future research.
In the present study, we present micromesh cultures as novel 3D cultures useful for cell maturation, microscopic observation, and integration into fluidic devices. When performing micromesh cultures, the mesh aperture size would be decided, depending on types of cells used or of experiments. Larger apertures have an advantage in microscopic analysis, and smaller apertures are suitable for cells with weak adhesiveness to mesh strands or other cells. We hope that these micromesh sheets are used as cell culture platforms for testing toxicity of drugs or finding new microphysiological phenomena in the future.
Footnotes
Acknowledgments
The authors thank Dr. Hiroo Iwata (RIKEN) and Mr. Nobutaka Tani (RIKEN) for valuable discussions and Dr. Shintaro Iwanaga (The University of Toyama), Dr. Kennedy Omondi Okeyo (Kyoto University), and Dr. Masao Washizu (The University of Tokyo) for experimental support. This work was supported, in part, by a grant-in-aid from the Hoyu Science Foundation (Aichi, Japan) and the Compass to Healthy Life Research Complex Program, RIKEN (Kobe, Japan).
Disclosure Statement
No potential conflicts of interest were disclosed.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.