
Abstract
Horses, like humans, can experience bone fractures and due to their large size and the need to bear weight on all limbs during the recovery period, they can be difficult to treat. Surgical techniques to improve fracture repair are improving, but to date, regenerative medicine technologies to aid fracture healing are not commonly applied in horses. We have previously demonstrated that equine induced pluripotent stem cells (iPSCs) can be differentiated into bone forming osteoblasts in 2D culture. In this study, we report on the use of a thermoplastic, 3D-printed polymer to provide a scaffold for successful, in vitro osteoblast differentiation of equine iPSCs. The scaffold provides a transparent, cost-effective solution to allow the analysis of osteoblast differentiation using live-cell imaging, immunohistochemistry, and quantitative polymerase chain reaction. This in vitro study demonstrates the future feasibility of generating 3D bone constructs through the cell seeding of scaffolds to use in regenerative medicine strategies to improve fracture repair in a relevant, large animal model.
Impact Statement
In this study, we describe the use of a cost-effective scaffold that can be used for in vitro studies of osteoblast differentiation by stem cells. The scaffolds can be printed to any size and shape, conditioned to improve cell adherence, and they are transparent to allow clear visualization of the cells in culture or postimmunohistochemical staining. Osteoblast differentiation of equine induced pluripotent stem cells was successfully performed and analyzed on a 3D-printed scaffold, which allows the future development of bone constructs to aid fracture repair in horses.
Introduction
Fractures caused by bone overloading or direct trauma are a significant welfare issue in multiple horse breeds taking part in a range of different disciplines. 1 Severe fracture leads to euthanasia, whereas smaller fractures can be treated conservatively with box rest and a cast. In delayed union or comminuted fractures, surgery is required, 2 but up to 40% of horses do not return to their previous athletic activity. 3 Regenerative medicine strategies using bone tissue engineering to improve fracture reunion and recovery would significantly improve horse welfare.
Bone grafts are used to promote bone regeneration and restore normal bone architecture in humans, 4 however, it is difficult to obtain sufficient tissue without donor site morbidity. In horses, autologous bone grafting has been performed for many years,5,6 but the effects on the donor site can be even more catastrophic, as the loss of tissue can lead to fracture at the donor site as the horse recovers from anaesthesia. 7 Using stem cells to enhance tissue healing is therefore becoming a popular alternative in both species.8–10
Induced pluripotent stem cells (iPSCs) are cells that are derived from somatic adult cells and have been reprogrammed such that they resemble an embryonic-like state and are capable of indefinite proliferation and can form cells from all three germ line lineages (endoderm, ectoderm, and mesoderm), 11 including bone-forming osteoblasts.12–14 We have successfully generated iPSC lines from equines 15 and have developed methods to differentiate the iPSCs into osteoblasts using traditional 2D cell culture techniques. 16 Equine iPSCs may therefore have the potential to provide large numbers of osteoblasts to utilize in tissue engineering strategies to aid fracture repair in horses.
It has been well documented that compared to 2D cultures, 3D systems provide more accurate modeling of the physiological and cellular environment of cells and promote and maintain lineage-specific differentiation and normal cellular architecture.17–19 The use of iPSCs with 3D scaffolds to enhance cell attachment, proliferation, and matrix deposition offer a promising option in regenerative medicine and allow cell organization that is more closely related to native tissues than 2D culture. 20 However, to date, there are no reports differentiation of equine iPSCs into osteoblasts on a 3D scaffold.
The overall aim of this study was to assess the potential of a 3D-printed polymer scaffold to support in vitro osteoblastic differentiation of equine iPSCs.
Materials and Methods
Thermoplastic 3D-printed polymers
Thermoplastic polymers Polycarbonate BendLay 3D Filament, transparent finish (Orbi-Tech) were printed using a Fused Deposition Modeling 3D printer (MakerBot Replicator 2 Desktop 3D Printer). The following printer settings were used: slicing setting was 0.2 mm, travel speed of the extrusion nozzle was 150 mm/s, z-axis speed was 23 mm/s, extruder temperature was 215°C, feeding rate of the filament was 30 mm/s, infill density was 100%, nozzle diameter was 0.4 mm, and the filament diameter was 1.77 mm. The pore size was set at 400 μm. The total thickness of the scaffolds was 1.66 mm, with each scaffold layer being 0.2 mm thick. Three different scaffold diameters were tested: 15.14, 14.90, and 14.60 mm. Two scaffold types were tested; open scaffolds and closed scaffolds. The only difference between them was the presence of a fine mesh (150 μm pore size) on the base of the closed scaffolds. Where present, a single layer of the fine mesh (0.2 mm thick) was printed as a continuation of the main scaffold.
The scaffold surface was conditioned by oxygen etching using a Bio-Rad PT7125 Barrel Plasma Etcher at 150 W, under 2 millibars pressure for 2 h to enhance cell adhesion. After printing, the scaffolds were preserved in a sealed container indefinitely at room temperature. After the oxygen etching, the scaffolds were preserved in a sealed container and stored at −20°C degrees until further use or at 4°C for up to 6 weeks.
Scanning electron microscope
The scaffolds seeded with 3T3 cells and cultured were fixed with 4% paraformaldehyde solutions in phosphate-buffered saline (PBS) overnight before gold coating on the sputter machine. The scaffold samples were loaded on to aluminum stubs with carbon tabs prefixed. The samples were gold coated using a Polaron SC7640 Sputter Coater, manufactured by Quorum Technologies. The coating parameters were 2.2 kV, 20 mA, 55 mm form Au target, and 30 s coating. The coated samples were washed three times with PBS and left to air dry before imaging on the Scanning Electron Microscope (SEM) machine for imaging (JSM 5900LV manufactured by JEOL fitted with a tungsten filament, acceleration voltage was set at 20 kV, and working distance was 12 mm).
Cell culture
Mouse 3T3 fibroblasts (from ATTC, Middlesex, United Kingdom) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 2 mM
Human osteosarcoma cell line Saos-2 cells (HTB-85 from ATTC) were cultured in 5% CO2, 37°C in McCoy's 5A medium with 15% fetal bovine serum, 2 mM
Six lines of previously derived equine iPSCs15,16 from three different horses were cultured on mitotically inactivated mouse embryonic fibroblasts in DMEM/F12, supplemented with 15% fetal calf serum, 2 mM
Bone differentiation on the constructs was carried out in osteoblast differentiation media. This consisted of the iPSC base medium (lacking bFGF and LIF) or the Saos-2 base medium supplemented with 10 mM β-glycerophosphate, 50 μM ascorbic acid, and 1 μM dexamethasone (all Sigma-Aldrich).
Before cell seeding, the constructs were sterilized under ultraviolet light (10 min/side) and conditioned with the cell-type appropriate media overnight. 1 × 10 4 cells were seeded onto each construct for all cell types. 3T3 and Saos-2 cells were seeded as single cells, and iPSCs were seeded as small colonies following mechanical passaging. Differentiation was carried out for 21 days with media replaced every 2–3 days.
Bone differentiation assays
To determine matrix mineralization, entire constructs were stained with von Kossa (Abcam, Cambridgeshire, United Kingdom) according to the manufacturer's instructions. Alizarin red S staining for calcium deposition was performed by incubating the entire constructs with 2% Alizarin red S pH 4.2 for 5 min. Hydroxyapatite deposition was detected using the OsteoImage bone mineralization assay (Lonza, Berkshire, United Kingdom) according to the manufacturer's instructions. Alkaline phosphatase (ALP) activity was measured using a quantitative colorimetric test on cell culture supernatant (Abcam) according to the manufacturer's instructions. Activity is measured in glycine units per milliliter where one glycine unit is the amount of enzyme causing the hydrolysis of 1 μmol of p-nitrophenyl phosphate per minute at pH 9.6 and 25°C.
RNA extraction, cDNA synthesis, and quantitative polymerase chain reaction
RNA was extracted using Tri-reagent (Sigma-Aldrich), purified using the RNeasy mini kit (Qiagen, Manchester, United Kingdom) and treated with Ambion DNA-free (Life Technologies, Paisley, United Kingdom). cDNA was made from 1 μg of RNA using the sensiFAST cDNA Synthesis Kit (Bioline, London, United Kingdom). Two microliters aliquots of cDNA were used in quantitative polymerase chain reaction (qPCR). Primers were designed using NCBI Primer-Blast (
Primer Sequences for Equine Gene Transcripts
Immunohistochemistry
This was performed on the entire constructs. The cells on the constructs were fixed in 3% paraformaldehyde for 20 min and permeabilized for 1 h with 0.1% Triton-X-100. They were washed in PBS and incubated with the primary antibodies overnight at 4°C before detection with an appropriate fluorescently labeled secondary antibody. All antibodies were used at optimized concentrations in PBS and appropriate negative controls were performed using secondary antibodies alone and IgG matched to the host species, as well as specific isotype of the primary antibody. Coverslips were mounted using VECTASHIELD HardSet Mounting Medium containing 4′,6-diamidino-2-phenylindole (Vector Laboratories, Cambridge, United Kingdom). Primary antibodies included mouse anti-collagen type I 1:100 (AB90395; Abcam), mouse anti-osteonectin (SPARC) 1:20 (MAB941-100; Bio-Techne, Oxford, United Kingdom), mouse anti-osteopontin (SPP1) 1:50 (21742; Santa Cruz Biotechnology, CA), rabbit anti-bone sialoprotein (IBSP) 1:100 (ORB1100; Biorbyt, Cambridge, United Kingdom), rabbit anti-RUNX2 1:50 (10758; Santa Cruz), goat anti-osteocalcin (BGALP) 1:50 (18319; Santa Cruz). Secondary antibodies were goat anti-mouse Alexa Fluor 594 1:200 (A11005; Invitrogen), goat anti-rabbit Alexa Fluor 594 1:200 (A11012; Invitrogen) and rabbit anti-goat Alexa Fluor 594 1:200 (Ab150144; Abcam). Constructs were inverted for imaging on a fluorescent microscope.
Results
Scaffold optimization
To fit into a standard 24-well tissue culture plate, the optimal size which allowed the insertion and removal of constructs from the well while minimizing the growth of cells under and around the construct was found to be 14.90 mm height, 14.90 mm width, and thickness 1.66 mm (Fig. 1).

The 3D scaffolds can be printed to fit into a standard 24-well tissue culture plate.
The surface coating was optimized to ensure the maximum adherence and growth of cells on the surface. Scanning EM revealed the adherence of 3T3 cells within the meshwork of the scaffold (Fig. 2A).
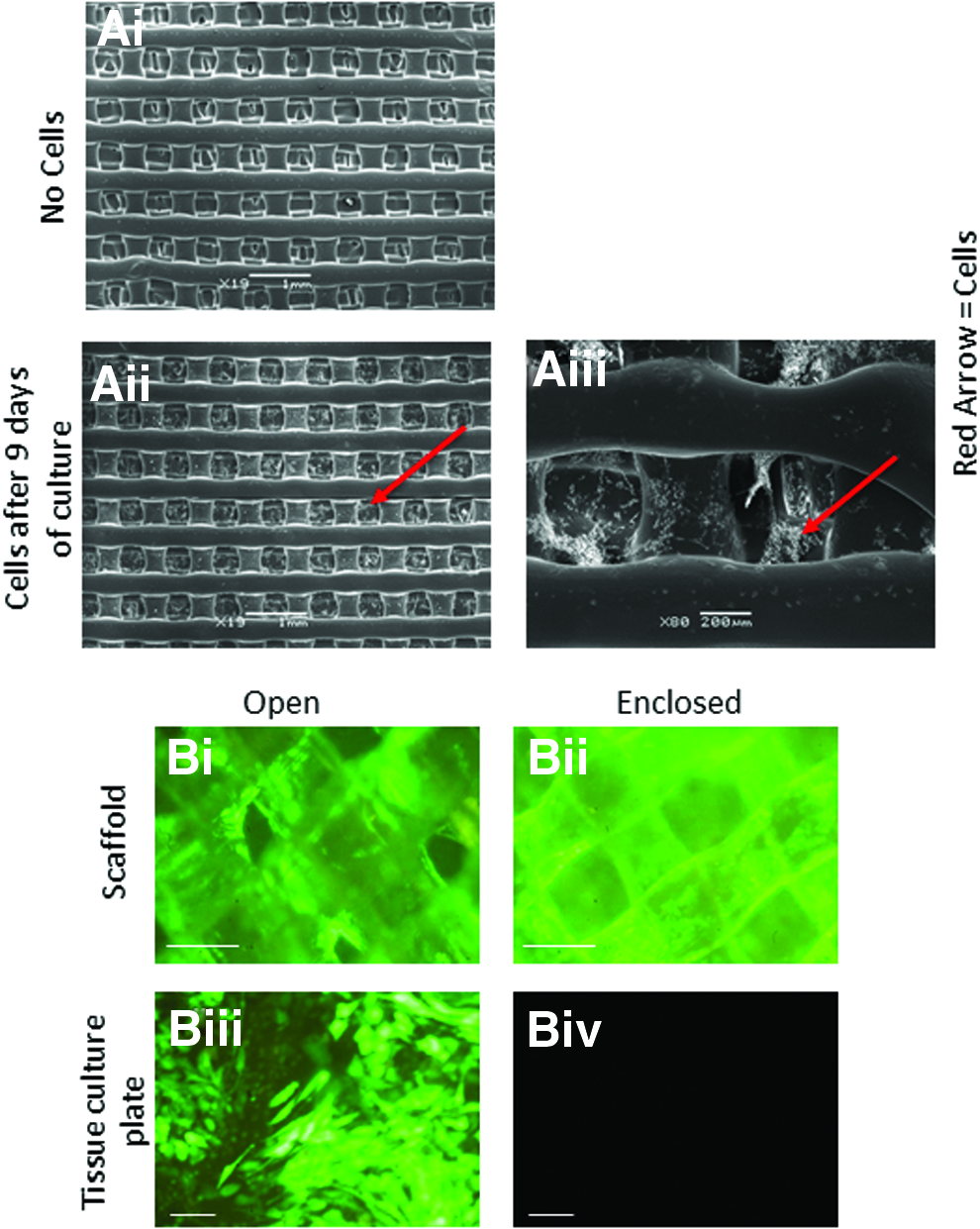
3T3 mouse fibroblasts and human Soas-2 cells can attach and proliferate to the 3D scaffolds.
Two types of construct were tested; open constructs and closed constructs. The closed constructs had a fine mesh layer that coated the bottom of the construct, enclosing it as a more isolated unit for cell growth, whereas the open construct lacked this fine mesh layer, meaning cells were free to penetrate through the construct and adhere to the bottom of the cell culture plate (Fig. 2B). Saos-2 cells expressing GFP were able to adhere to the scaffold of both construct types, but the open constructs with no mesh had far fewer cells remaining within the construct and many cells present on the bottom of the culture dish. In contrast, the enclosed construct did not allow any cells to pass through to the bottom of the culture dish. Enclosed constructs were therefore used in all further experiments.
iPSCs and Soas-2 cells differentiated on the 3D scaffold produce a mineralized matrix
GFP-labeled equine iPSCs were seeded as small colonies onto the constructs. These adhered to the constructs and individual cells migrated away from the colonies along the scaffold, but after 21 days of differentiation the iPSC-derived cells were fewer in number and less evenly distributed than Saos-2 cells (Fig. 3). Despite the ability of the cells to move through the pores, cells were attached throughout the depth of the scaffold (rather than just on the base of the enclosed construct). This can be seen in Figure 3 where the cells are clearly present on the scaffold layers containing pores of 400 μm.
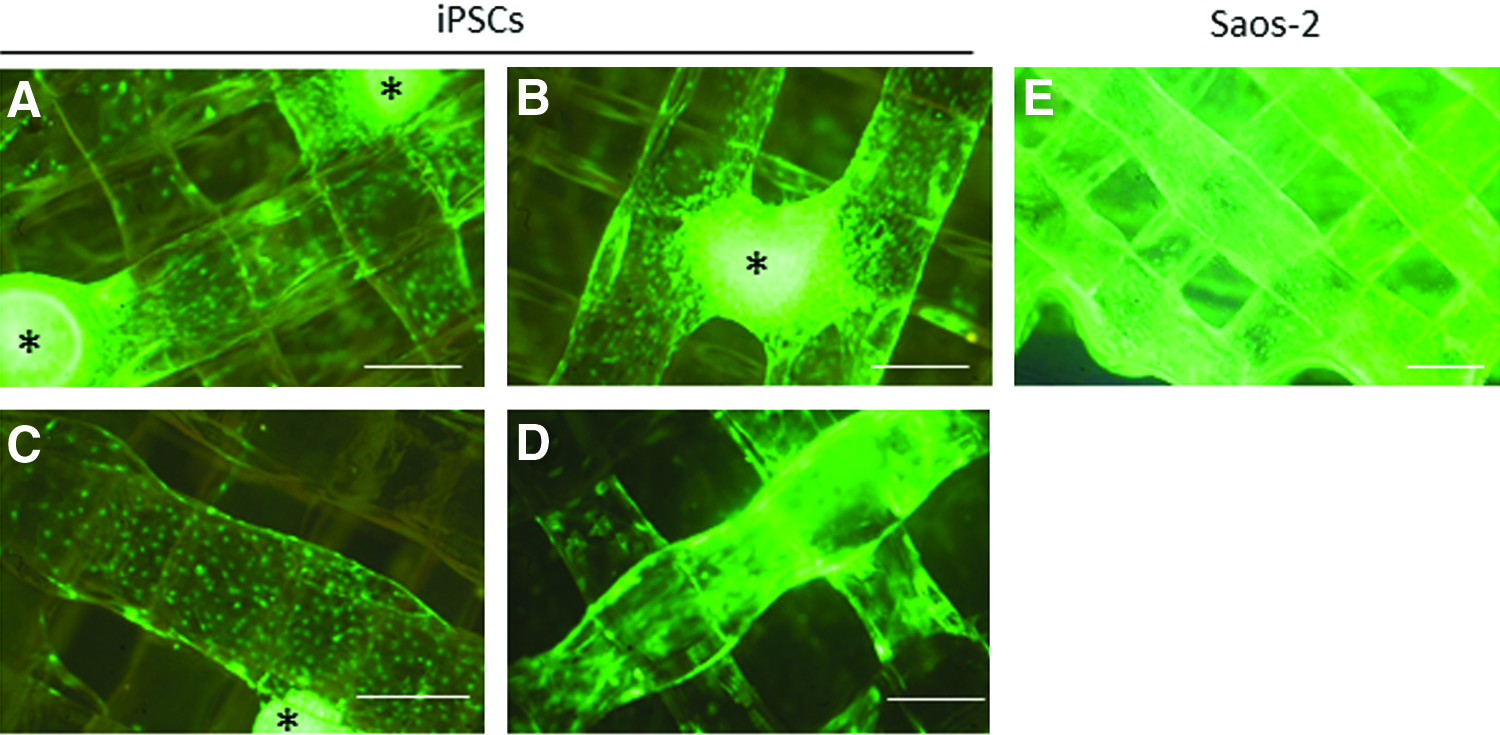
GFP labeled iPSCs seeded as colonies and differentiated for 21 days on the 3D scaffold. iPSC colonies (indicated by *) adhere to the scaffold and cells migrate out along the scaffold fibers
Alizarin red S and von Kossa staining on the entire constructs demonstrated that calcium had been deposited by both Saos-2 and iPSCs following 21 days of culture in osteoblast differentiation medium (Fig. 4A, B). More intense and global staining of constructs seeded with Saos-2 cells was observed than in constructs seeded with iPSCs. Control constructs, in which no cells had been seeded, did not produce any positive staining.

iPSCs differentiated on the 3D scaffold produce a mineralized matrix.
Non-GFP labeled iPSCs were seeded onto constructs in osteoblast differentiation media for 21 days before fluorescent detection of hydroxyapatite. Clear deposition of hydroxyapatite could be visualized on the scaffold (Fig. 4C). All Saos-2 cells used in these 3D studies were labeled with GFP, and therefore, we were not able perform fluorescent detection of hydroxyapatite on the Saos-2 seeded scaffolds.
iPSCs and Soas-2 cells differentiated on the 3D scaffold synthesize ALP
A low level of ALP activity was produced by undifferentiated iPSCs, but the level of ALP activity increased over the 21 days of differentiation by 20.5-fold (Fig. 5). This was still lower than the level of ALP activity for the Saos-2 cells, which was 1.5-fold higher than for the iPSCs after 21 days of differentiation.
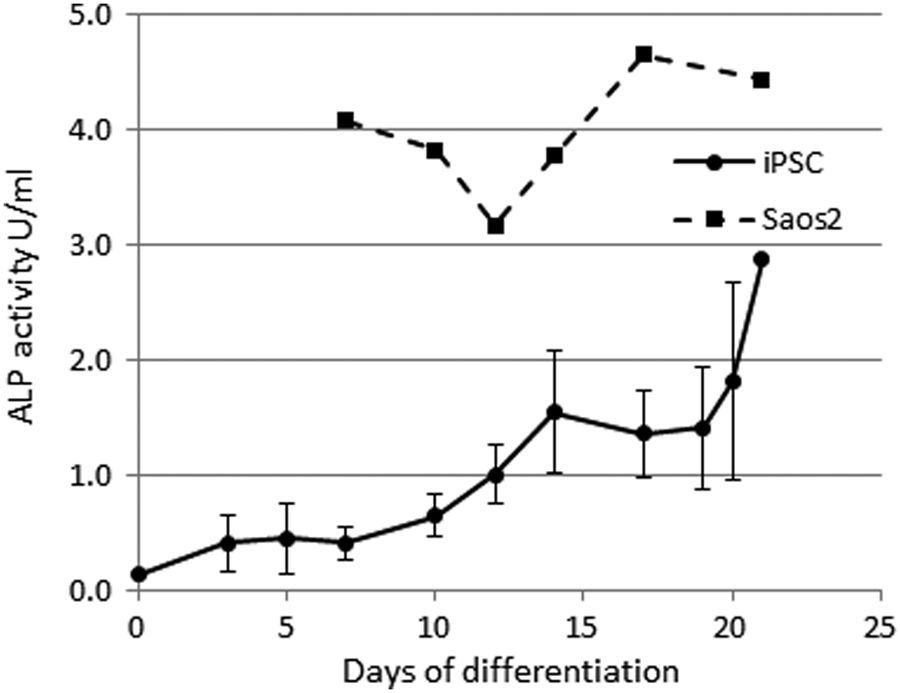
iPSCs differentiated on the 3D scaffold have increasing ALP activity with time. Error bars represent the standard error of the mean from a total of six clonal lines of iPSCs derived from three different horses. ALP from one replicate of Saos-2 cells differentiated on the 3D scaffold was measured as a positive control. ALP, alkaline phosphatase.
iPSCs differentiated on the 3D scaffold express osteoblast-associated genes and proteins
After 21 days of differentiation on the 3D scaffolds, the iPSCs expressed the osteoblast-associated genes COL1A1 (collagen type I), SPARC (osteonectin), RUNX2 (runt-related transcription factor 2), SPP1 (osteopontin), and BGLAP (osteocalcin), but no expression of IBSP (integrin-binding sialoprotein) was detected (Fig. 6). Similarly, immunohistochemical staining for osteoblast-associated proteins revealed positive, cell-associated staining for COL1A1, SPARC, RUNX2, SPP1, and BGALP, but not IBSP (Fig. 6). Gene and protein expression for the Saos-2 was not examined in this study.
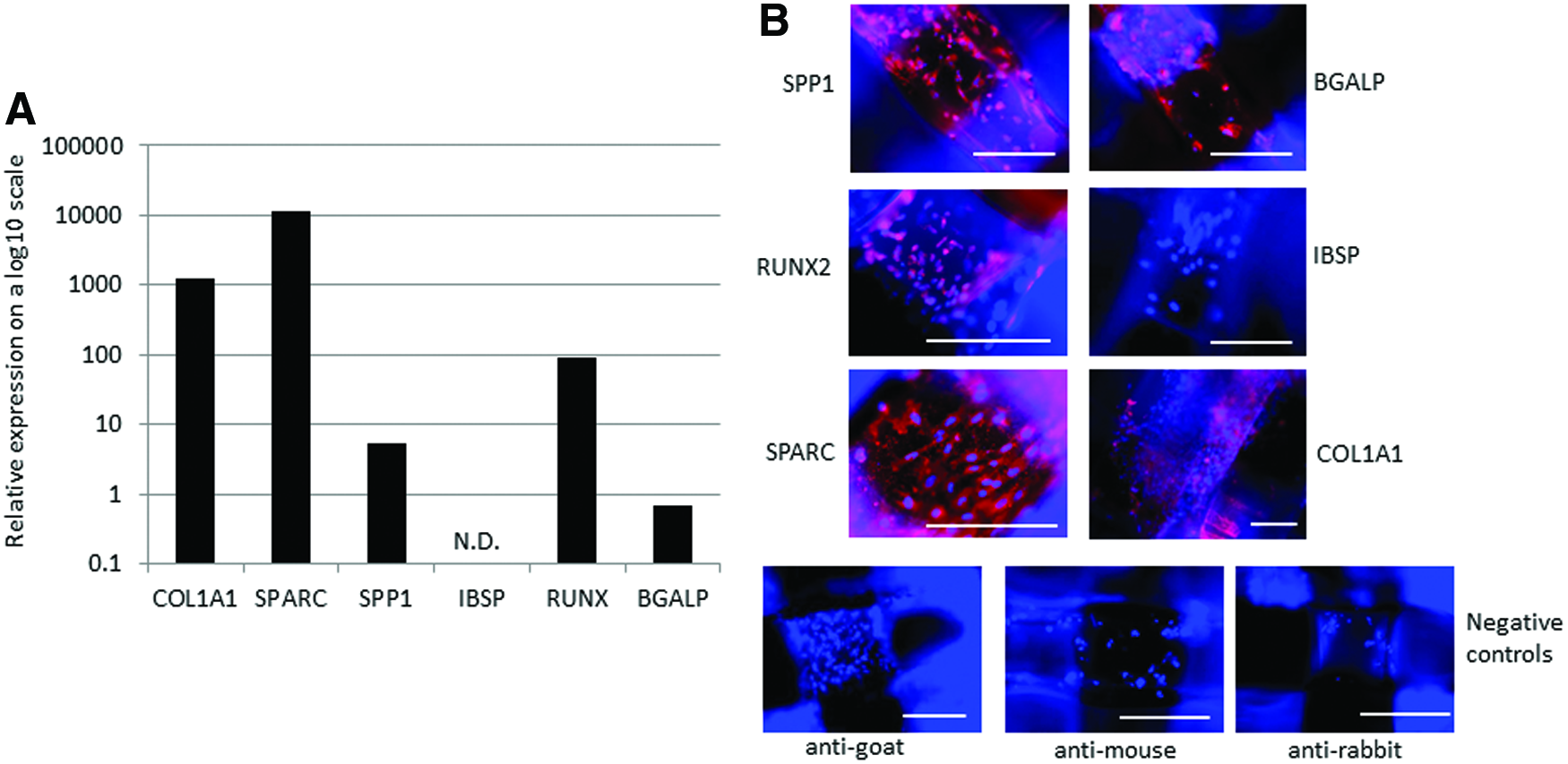
iPSCs differentiated on the 3D scaffolds for 21 days express osteoblast-associated genes and proteins.
Discussion
We have previously reported the successful 2D differentiation of equine iPSCs into osteoblasts, 16 and in this study, we report the use of a novel, thermoplastic 3D-printed scaffold to allow in vitro 3D osteoblast differentiation of equine iPSCs.
The filament used to make the scaffolds is biocompatible, 22 cheap to purchase, and can be printed to any size or shape. Importantly, it is also optically transparent, whereas other printable polymers such as polylactide (PLA) and polycaprolactone are opaque. Here, we optimized the scaffold fit to a standard 24-well tissue culture plate and performed oxygen etching to allow good adhesion of the cell types being tested. Without surface treatment, cells do not attach to the BendLay polymer. The polymer can be coated with other proteins such as fibronectin, collagen type I, and laminin to promote cell adhesion (unpublished data), but we have only determined equine iPSC attachment and differentiation on oxygen-etched polymers. This is a similar process to that performed in the manufacture of tissue culture plastics. We found that enclosed constructs containing a fine mesh layer on the base, retained the cells within the scaffold much more effectively than open scaffolds lacking the mesh layer, where many cells were found adhered to the bottom of the cell culture well. We did not test the use of ultralow attachment culture plates in this work, but if open scaffolds are required (for example to improve blood vessel infiltration in vivo), low attachment plates may help encourage cell retention on the scaffolds rather than on the culture plate itself. Pores within the scaffold allow the diffusion of nutrients and cellular proliferation, migration, and communication. A wide range of pore sizes have been used in bone tissue engineering, ranging from 20 to 1500 μm, 23 and in this study, we demonstrated that a combination of 150 and 400 μm provided an effective scaffold for in vitro differentiation and analysis.
In this study, we used scaffolds which were 1.66 mm thick and found that we had very good distribution of the cells throughout the depth of the scaffold. However, future work to determine the effect of scaffold thickness on cell survival, migration, and differentiation is required as the diffusion of nutrients may become limited as the thickness increases.
The transparency of the scaffold is of great benefit for in vitro studies, as it allows the distribution and growth of the cells to be visualized during standard culture. How cells are distributed, proliferate, and differentiate on a scaffold can effect the likely functionality of the engineered tissue, 24 and assessing this on nontransparent scaffolds requires additional techniques to be performed. 25 Furthermore, transparent scaffolds enable the use of the same protocols for the end point analysis of differentiation to be conducted as for 2D differentiation (e.g., immunocytochemical staining). Therefore, as a tool for studying in vitro bone differentiation, this scaffold provides a cost-effective option that allows robust postdifferentiation analyses to be performed.
Saos-2 cells seeded onto the constructs as single cells exhibited a very even distribution throughout the scaffold after 21 days of culture, whereas equine iPSCs, seeded as small colonies, were less well distributed after 21 days of culture, and future work to enable the seeding of the iPSCs as single cells would likely be beneficial. Nevertheless, iPSCs did grow out of the original colonies along the meshwork of the scaffold. However, we were unable to find a successful method for extracting live cells from the scaffold to determine actual cell numbers at the end of the 21 days of differentiation. The extraction of live cells is likely complicated by the fact that upon differentiation into osteoblasts, the cells produce a mineralized matrix. 16
We used both Alizarin red S and von Kossa staining to detect calcium production by the differentiated iPSCs and acknowledge that von Kossa reacts with the anionic portion of many salts and is not specific for calcium. 26 The iPSC-seeded constructs demonstrated distinct patches of calcium deposition, which may correlate with areas containing more cells, indicating successful osteoblast differentiation. Saos-2 seeded constructs were used as a positive control and had much more observable Alizarin red S and von Kossa staining, which was evenly distributed across the constructs. This likely reflects the even distribution of the Saos-2 cells and the fact that as a human osteosarcoma cell line, they readily undergo osteoblast differentiation to produce a mineralized matrix. 27 Non-GFP labeled equine iPSCs were also seeded on constructs and used to detect hydroxyapatite. Clear deposition of hydroxyapatite was visible on the meshwork of the scaffold supporting conclusion that the iPSCs had differentiated into osteoblasts.
Bone mineralized requires the activity of ALP, 28 and we demonstrated that over the 21 days of differentiation there is an increase ALP activity. As we reported previously, 16 and similar to human and mouse embryonic stem cells, 29 undifferentiated equine iPSCs express a low level of ALP. ALP activity is increased ∼20.5-fold after 21 days of differentiation in 3D. In comparison, we previously reported that ALP activity is only increased ∼6-fold after 21 days of 2D differentiation, 16 but we have not performed these experiments in parallel. ALP activity was only measured in one replicate of Saos-2 differentiation, and so, it was not possible to perform a statistical analysis, however, the level of ALP activity was higher in differentiating Saos-2 cells than iPSCs.
Quantitative PCR and immunohistochemistry produced matching results and demonstrated the expression of COL1A1, SPARC, SPP1, RUNX2, and BGALP by the differentiated equine iPSCs at both the gene and the protein level. Of note, the iPSCs that had grown out of the original colonies and existed as single cells on the scaffold were positive for the bone protein markers. This demonstrates that the migrating cells do undergo differentiation. However, no gene or protein expression was detected for IBSP. We have previously demonstrated that IBSP expression following 2D differentiation of equine iPSCs shows large amounts of inter-horse variability. 16 However, it may also reflect the fact that IBSP (bone sialoprotein) is a later marker of osteoblastic differentiation than the others that were tested.30,31 RUNX2 is a key transcription factor in the osteoblast differentiation pathway32,33 and has a relatively high expression after 21 days of iPSC differentiation. Similarly, COL1A1 and SPARC have relatively high gene expression levels. These are upregulated in the early phases of differentiation34,35 and are considered to be early markers of osteoblast differentiation.36–38 In contrast, BGALP and SPP1 have lower expression levels and are both required in the later stages of bone formation.36,39,40 Although these analyses were only performed on a single line of iPSCs and further replicates are required, they do suggest that the iPSCs may require additional time in culture to generate a more mature osteoblast phenotype. Alternatively, as the material used in the scaffold has been shown to affect the efficiency of bone differentiation of a variety of cell types,17,41,42 coating the scaffold with osteogenic factors may lead to more efficient differentiation of the iPSCs.
Improving the efficiency of differentiation of iPSCs is particularly important with regard to their clinical application because any remaining undifferentiated cells may have the potential to undergo uncontrolled proliferation and tumor formation in vivo. 43 As we were not able to extract live cells from the scaffold, it was not possible to quantify the efficiency of differentiation of the iPSCs, for example using flow cytometry. Other methods using an intermediate differentiation step to mesenchymal stem cells 44 and removal of any undifferentiated iPSCs (using cell sorting) before 3D differentiation may help to reduce the safety concerns of using iPSC-derived products clinically. Future work to determine the optimal duration of 3D differentiation is also required, as in this study, we only examined differentiation after 21 days.
These polycarbonate scaffolds are not biodegradable and would not break down over time in vivo in line with tissue healing. However, many nonbiodegradable scaffolds/implants are used in in vivo fracture repair such as metals and PAA (polyacrylic acid) 45 and generally biodegradable scaffolds have poorer mechanical properties that can make them unsuitable for fracture repair in bones exposed to large forces.45,46 The polycarbonate scaffolds used in this report can be easily scaled up and printed to any size or shape and can be fabricated rapidly. This would allow them to be custom produced to fit individual bone defects, while ensuring that the material, pore size, and so on remain standardized. Polycarbonate is lightweight, but has a high tensile strength of 77 MPa and a high tensile break strength of 75–150%, which means that it can withstand high loads and torsional stress far better than other thermoplastics, which have been used in bone repair such as PLA 47 and polymethyl methacrylate. 48 This is of particular importance for equine clinical applications where limb bones are exposed to high loads, and the native tissue has a high ultimate tensile strength of around 120 MPa. 49
In summary, we report that equine iPSCs can be successfully differentiated into bone-forming osteoblasts on a thermoplastic, 3D-printed polymer, which form the development of novel methods for improving fracture repair in horses in the future.
Footnotes
Acknowledgments
The authors thank The Petplan Charitable Trust for funding this work (Project No. 227-265). A. Baird was kindly funded by the Paul Mellon Foundation and Anne Duchess of Westminster Charitable Trust.
Disclosure Statement
No competing financial interests exist.