
Abstract
Most commonly used cell lines are readily susceptible to genome editing and present a good object for cell models to establish disease-causing genes and find ways to cure diseases. However, karyotype and phenotype heterogeneity between individual cells in such cultures as well as multiplicity of target alleles make generation of desired cell lines by single-cell cloning (used for diploid cells) inapplicable. We designed and tested a simple approach for targeted genome modification of single cells in sizable cell populations, containing multiple karyotype and phenotype variants. To obtain the cell lines with suppressed expression of target proteins, we applied an original multiround genome modification protocol, monitoring protein expression level and impairment of target and off-target (undesired) DNA cleavage sites. We found that repeated modifications increase efficacy of target DNA allele disruption and decrease expression of corresponding proteins in cell populations in vitro. However, certain off-target activity was observed as well. Unexpectedly, we did not detect the increment of de novo off-target DNA site cleavage after CRISPR/Cas9 reuse, which proves our approach is suitable for genome editing in aneuploidy cell lines. Our protocol can be used for in vitro model creation by genome editing of aneuploid cells or cells with restricted clonogenic potential.
Impact Statement
Cell lines represent convenient models to elucidate specific causes of multigenetic and pluricausal diseases, to test breakthrough regenerative technologies. Most commonly used cell lines surpass diploid cells in their accessibility for delivery of large DNA molecules and genome editing, but the main obstacles for obtaining cell models with knockout-targeted protein from aneuploid cells are multiple allele copies and karyotype/phenotype heterogeneity. In the study, we report an original approach to CRISPR-/Cas9-mediated genome modification of aneuploid cell cultures to create functional cell models, achieving highly efficient targeted protein knockout and avoiding “clonal effect” (for the first time to our knowledge).
Introduction
Cell lines represent convenient models to study complex biological objects, to elucidate specific causes of multigenetic and pluricausal diseases and to test breakthrough regenerative technologies.1,2 Genome editing increases potential of cell models because it allows to modify genome sequences in a specific manner (knockout, knock-in, single nucleotide polymorphism [SNP] modeling, etc.). However most widely used cell lines (e.g., HEK293, NIH/3T3, HeLa, HepG2, and Neuro2a) have unstable aneuploid karyotype and, thus, cell populations show highly heterogeneous genomes and phenotypes.3–5
Because of aneuploidy, each cell in such population has a personal “dosage” of each chromosome and off-target allele as well (typically, from 0 to 6). 3 Multiple copies of targeted alleles and great variability of clones' characteristics make an approach used for genome editing of diploid cell cultures (with stable chromosome numbers) inapplicable for genome modification of aneuploid cells. Obtained clones would not inherit the properties of the parental cell culture (the so-called, “clonal effect”) 6 ; therefore their progenies are not suitable for study of complex cellular processes: intracellular signaling, proliferation, migration, regeneration, and so on. Moreover, it would be challenging to knockout multiple target alleles during a single round of modification, considering moderate CRISPR/Cas9 efficiency (from 30% to 60% per allele). However, most transformed cell lines surpass diploid cells in their accessibility for delivery of large DNA molecules and genome editing, so they are still a very attractive object for creation of model systems. Still, to our knowledge, there are no works in which populations (not clones) of aneuploid cells with edited genome were obtained and functionally tested.
In our previous work, we applied genome editing protocol for diploid cells with subsequent single-cell cloning to obtain clones of aneuploid cell lines (HeLa, Neuro2a, etc.) with knocked-out target genes, yet we encountered above-mentioned hurdles namely “clonal effect” and multiple of target alleles. We supposed that it would be possible to suppress target protein expression in general cell culture that contains numerous karyotype and phenotype variations. To prove our hypothesis and to obtain desired cell lines, we applied a multiround genome editing protocol. The idea to use CRISPR/Cas9 in one cell culture repeatedly (for increasing its efficacy) is evident, but no one knows if this approach is more effective or less accurate. In this study, we show that it is suitable for creation of functional cell models and prove it by monitoring protein expression, disruption of target DNA loci, and controlling undesired DNA cleavage sites.
Materials and Methods
Cell cultures
The choice of genes for knocking out was dictated by the needs of our laboratory. We studied various aspects of regenerative processes, so molecules associated with cell migration (CDH13 and PLAUR), inflammation (DUOX1/2 and NOX4), and differentiation (HHEX) were knocked out. The choice of cell lines (Table 1) was determined by their availability and functionality of the molecules of interest in them.
Cell Lines and Targeted Genes
GPI, glycosylphosphatidylinositol.
Cells were cultured in Dulbecco's Modified Eagles Medium (DMEM) High Glucose (HyClone; #SH30022FS) supplemented with 10% of fetal bovine serum (HyClone; #SH3007103) and 1% Penicillin-Streptomycin-Glutamine solution (ThermoFisher Scientific; #10378016). All cell cultures were passaged at a dilution 1:3–1:5, using HyQtase solution (HyClone; #SV3003001) before they reached confluency.
Experimental design
After we found that progenies of individual clones vary dramatically in their characteristics and behavior and that single-round modification was not enough to knock out target protein expression in pooled cell populations (Fig. 1 and Supplementary Figs. S1–S4), we designed the following approach (Fig. 2B): with the help of Lipofectamine2000 reagent (ThermoFisher Scientific; #12566014), we transformed the cell population with pX458 plasmid (Addgene; #48138), encoding Cas9 (CRISPR-associated nuclease), green fluorescent protein (GFP), and an appropriate or no guidance RNA (gRNA), according to the recommendations of the manufacturer. About 48 h later, we sorted GFP bright positive cells using fluorescence activated cell sorting (FACS) and cultivated them for 7–10 days until GFP fluorescence faded. Then, we repeated modification-sorting-cultivation procedure twice more. After each modification round, samples of sorted cell populations were stored and single-cell clones were obtained from some of them. These pooled populations and individual clones were tested for target protein expression (Western blotting [WB], flow cytometry) (n = 3) and for disruption of on-target and off-target DNA sites (Sanger sequencing) (n ≥ 1). Selected cell populations were used for further functional tests (n ≥ 3).
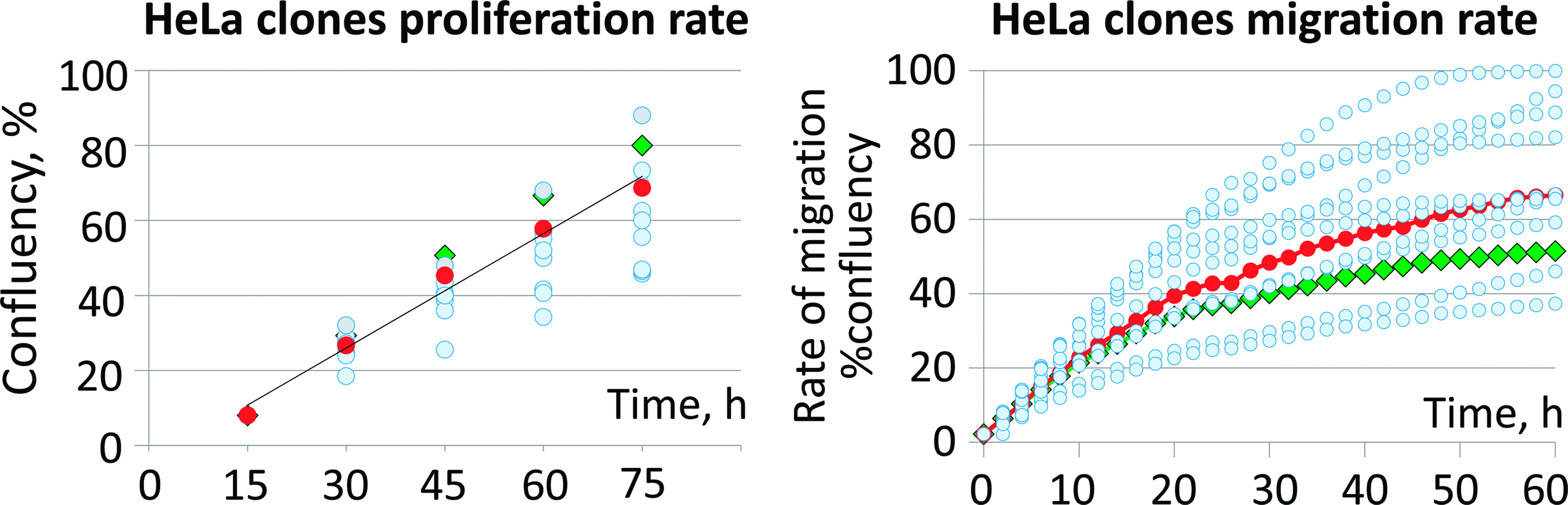
HeLa clone proliferation and migration rates (circles). Mean values are given (n = 8). Triangles—initial HeLa population; diamonds—artificial HeLa population. Color images are available online.

Comparison of conventional protocol for gene knockout in diploid cells
Flow cytometry and cell sorting
Cells were detached from culture dish and resuspended in sterile phosphate-buffered saline (PBS) with 1% bovine serum albumin (BSA). For flow cytometry analysis, the cells were stained for 30 min at room temperature using primary antibodies, washed, and restained for 30 min using appropriate secondary antibodies, coupled to fluorochromes. For CDH13 staining, Cadherin-13 antibody (LSBio; #LS-C35237-100) and F(ab)2 IgG APC-conjugated antibody (R&D; #F0113) were used in dilution 1:20. For PLAUR staining, uPAR antibody (Santa Cruz; #SC-10815, dilution 1:50) and FITC anti-IgG Fc antibody (Biolegend; #409310, dilution 1:20) were used. As isotype control Rat and Rabbit IgG (Invitrogen; #02-9602 and #10500C, dilution 1:500) were used as primary antibodies. Flow cytometry was performed on LSR Fortessa instrument (BD Biosciences).
FACS was performed on BD FACSAria III instrument (BD Biosciences). Cells resuspended in PBS with 1% BSA were sorted on the basis of GFP emission intensity. GFP bright sorted cells were collected directly to 24-well plates and cultured for 7–10 days before analysis or further modification. For clonal analysis, single cells were sorted separately in wells of a 96-well plate. HeLa single-cell clones used in proliferation and migration tests were obtained by direct sorting of unstained (unmodified) cells into the wells of a 96-well plate.
Measurement of cell proliferation and migration
To assess the rate of proliferation, HeLa clones were plated in a 96-well plate (5 × 10 3 cells per well) and placed in the Incucyte® ZOOM Live Cell Analysis System (Essen Bioscience). Cell proliferation was measured every 5 h using live cell time-lapse imaging with following analysis of the obtained data with Incucyte ZOOM image processing software package. The experiment lasted until the first clones formed a monolayer—it took about 3–4 days.
The IncuCyte ZOOM Scratch Wound assay was performed using WoundMaker ImageLock Plate (Essen Bioscience). Progenies of HeLa clones were plated in a 96-well plate (5 × 10 4 cells per well) (n = 8), grown to a monolayer, and then a scratch was made using the WoundMaker Plate. Cellular debris was washed and the plate was placed in Incucyte. Cell migration was assessed every 2 h using live cell time-lapse imaging. Evaluation of Wound Confluence parameters was performed using the IncuCyte Scratch Wound Cell Migration Software Module.
gRNA design
One of the tasks of this study was to obtain cell lines with appropriate genes knocked out or edited—and this determined gRNA design, performed using Cas-Designer web tool. 9 All gRNAs target exonic sequence of appropriate genes and are designed to knock out by reading frameshift, except Duox2 gene. Its gRNA targets DNA sequence, corresponding C-terminus of DUOX2 protein (last 23 amino acids). Thus, we tried to disrupt only certain activities of multifunctional DUOX2 protein.
Among the proposed gRNAs, we selected those with minimal expectancy of off-target activity. Corresponding DNA sequences were synthesized as oligonucleotides and cloned as DNA-duplexes to BbsI restriction sites of pX458 or pX458nickase vectors according to the previously described protocol. 10 pX458nickase (D10A) vector was generated by site-directed mutagenesis. Sanger sequencing of the resulting plasmids confirmed the fact of integration of the DNA sequences, encoding gRNAs' spacers. Two most likely off-targets were predicted by COSMID web tool. 11 Primers for PCR amplification were designed using Primer-Blast web tool. 12 The sequences of gRNAs' spacers, potential off-target DNA sites, and oligonucleotides used for PCR amplification are listed in Supplementary Table S1.
CRISPR/Cas9 genome modification of the cells was performed by lipofection of cell populations with pX458 plasmid as described above. To obtain control groups for functional studies, some portions of appropriate cell lines were transfected with unmodified pX458 plasmid.
Western blotting of cell lysates
Polyacrylamide gel electrophoresis and Western blotting were performed according to the standard protocol. 13 For target protein detection, the following primary antibodies were used: CDH13 (Abcam; #ab167407, dilution 1:3000), PLAUR (Santa Cruz; #SC-10815, dilution 1:500), HHEX (Abcam; #34222, dilution 1:500), DUOX1 (Novus Biologicals; NBP2-16232, dilution 1:1000), DUOX2 (Novus Biologicals; NB110-61576, dilution 1:500), NOX4 (Novus Biologicals; NB110-58849, dilution 1:500), ACTB (GeneTex; #GT5512, dilution 1:2000), TUBB3 (BioLegend; #MMS-435P, dilution 1:500–1:1000), and VCL (Abcam; #ab91459, dilution 1:2000). Membranes stained with primary antibodies were washed and restained in the solution of appropriate secondary antibody conjugated with horseradish peroxidase. To develop the signal, SuperSignal™ West Pico PLUS Chemiluminescent Substrate (ThermoFisher Scientific; #34580) was applied onto the blot paper. The signal was detected by Chemi Doc XRS (BioRad) and processed with Image Lab 3.0 (BioRad).
Sanger sequencing of DNA On- and Off-targets
DNA from clones and cell populations was extracted. DNA fragments containing sites of on-target and potential off-target Cas9-mediated cleavage were PCR amplified using primers listed in Supplementary Table S1. PCR fragments were cleared by agarose gel electrophoresis, isolated from agarose gel, and sequenced (Evrogen). Sequencing data were analyzed using TIDE web tool 14 and ChromasLite 2.1.1. The minimal number of alleles per single-cell clone was estimated by DNA sequence desynchronization in the way similar to a previously described method that used SNPs. 15
Results
Migration and proliferation rates differ dramatically between HeLa clones
HeLa cell line was modified (targeted gene CDH13) before others. After its modification, we found that single-cell clones' progenies proliferate and migrate diversely, and in the control group (no gRNA) as well. This could not be explained by genome modification effect, so we supposed it could be the consequence of “clonal effect.” To prove it, HeLa cell line was single-cell cloned to obtain 10 clones and their progenies with further assessment of their migration and proliferation. Cells' proliferation and migration depend on numerous biochemical reactions and signaling cascades, so we used these integrative characteristics as a measure of cell “well-being.” We found that cultures obtained from HeLa single-cell clones differ dramatically in their proliferation and migration rates (Fig. 1, blue circles). The most likely causes of this difference are the diverse combinations and dosages of chromosomes between the ancestor clones. The initial HeLa population due to diversity of chromosome combinations should display medial level of proliferation and migration and our observation confirmed that (Fig. 1, red circles). The artificial HeLa population obtained as a mixture of progenies of 10 single-cell clones migrates and proliferates slightly different from initial HeLa population (Fig. 1, green diamonds). This forced us to choose a genome editing protocol for pooled cell population, not for single-cell clones.
Multiround modification increases efficacy of genome editing in aneuploid cell populations
We tried to obtain HeLa cell line with knocked-out CDH13 gene after a single round of modification; however, we found that used approach was not efficient enough (Fig. 3 and Supplementary Figs. S5 and S6 “HeLa_s1”) and we switched to using the multiround modification protocol. Thus, HeLa and other cell lines passed through three modification rounds—each consisted of genome editing procedure followed by sorting of GFP-positive cells. We found that after each round, the level of expression of targeted proteins decreased as detected by WB and flow cytometry (for surface-localized CDH13 and PLAUR) (Fig. 3 and Supplementary Figs. S5–S7). However, efficacy of targeted protein suppression varied between cell populations and target genes. As for Duox2 gene, it was not knocked out—just its C-terminus (last 23 amino acids) was disrupted by reading frame shift. It did not change DUOX2 molecular weight significantly, neither destroyed the epitopes recognized by DUOX2-specific antibodies, so we did not observe any change in DUOX2 expression after the third round of modification (Fig. 3 and Supplementary Figs. S5 and S6).
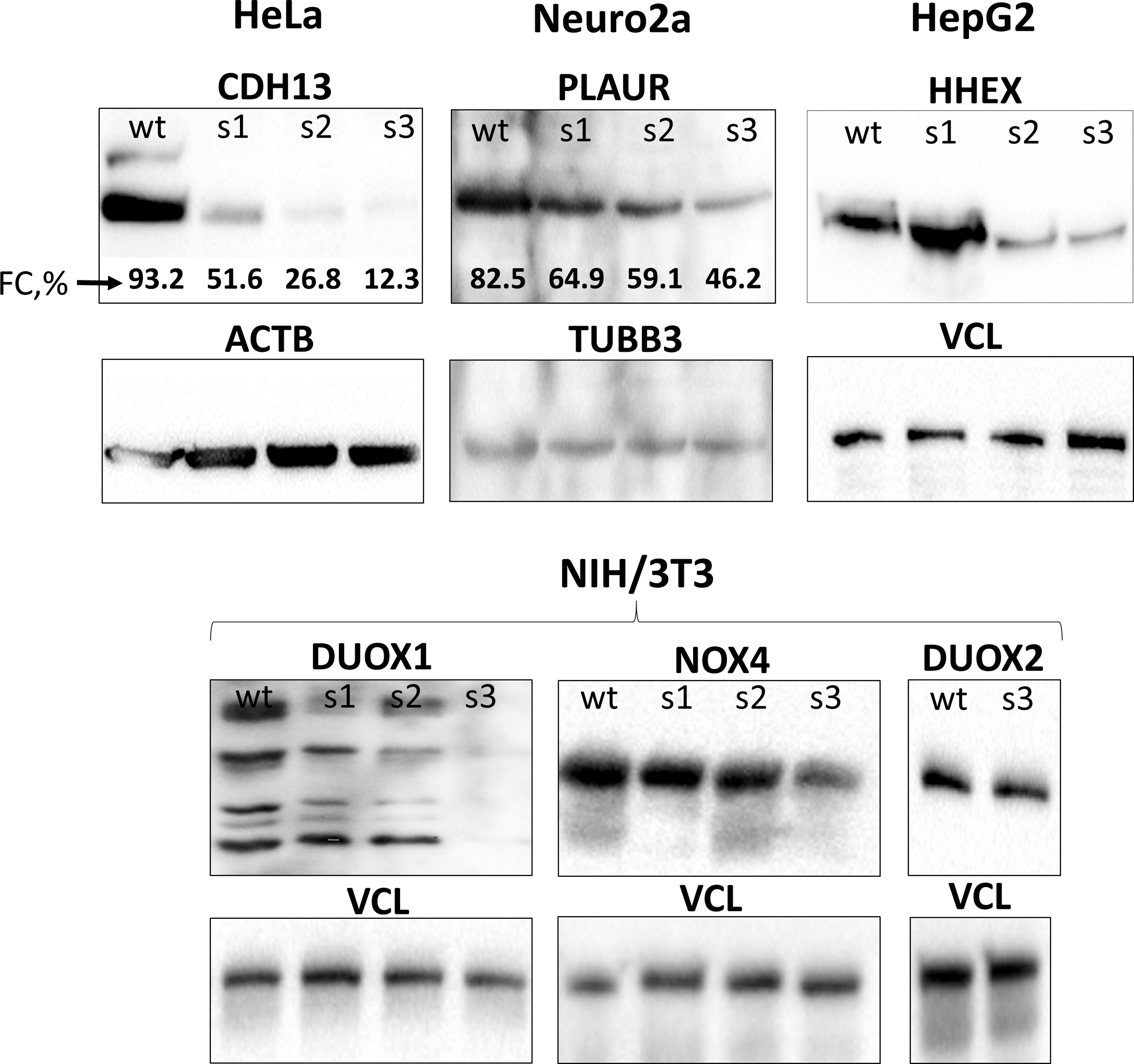
Dynamics of target protein (CDH13, PLAUR, HHEX, DUOX1, NOX4, and DUOX2) expression in cell populations after each round of modification assayed by WB and flow cytometry (FC, %—percentage of stained cells, Supplementary Fig. S7). wt, wild type; s1, s2, and s3—pooled populations for each consecutive round of modification; ACTB, beta-actin; TUBB3, beta3-tubulin; VCL, vinculin; WB, Western blotting.
Obtained data were confirmed in analysis of single-cell clones' progenies of “HeLa_s3,” “Neuro2a_s3,” and “NIH/3T3_Nox4_s3” cell lines (Fig. 4 and Supplementary Figs. S8 and S9). Majority of analyzed clones (especially in “HeLa_s3” population) did not express target proteins at detectable levels according to WB (Fig. 4 and Supplementary Figs. S8 and S9). Pursuant to the results of genome sequencing, the overwhelming majority of target alleles in those clones were successfully edited. Number of CDH13 alleles varies from clone to clone (from 1 to 3). Some alleles restored the reading frame due to deletions of nucleotide multiple of three (Supplementary Table S2–S4), but still they did not provide CDH13 expression (Fig. 4 and Supplementary Figs. S8 and S9 “HeLa_s3”).

Target protein (CDH13, PLAUR, and NOX4) expression in progenies of single-cell clones of “HeLa_s3,” “Neuro2a_s3,” and “NIH/3T3_Nox4_s3” populations after the third round of modification (WB). C2, C3, etc., clones #2, #3, etc.
The analysis of on-target and off-target DNA sites in pooled cell populations after each modification round demonstrated that the degree of damage to on-target sites increases from round to round. At the same time, cleavage of potential off-target DNA sites seems to happen very rare in second and third rounds if they have not been cleaved during the first modification round. In several cases, off-target DNA sites were edited during the first modification round, but observed that off-target CRISPR/Cas9 activity was less prominent than on-target (Fig. 5). These results were confirmed by Sanger sequencing of on-target /off-target DNA sites of single-cell clones' progenies (Supplementary Table S2–S5).
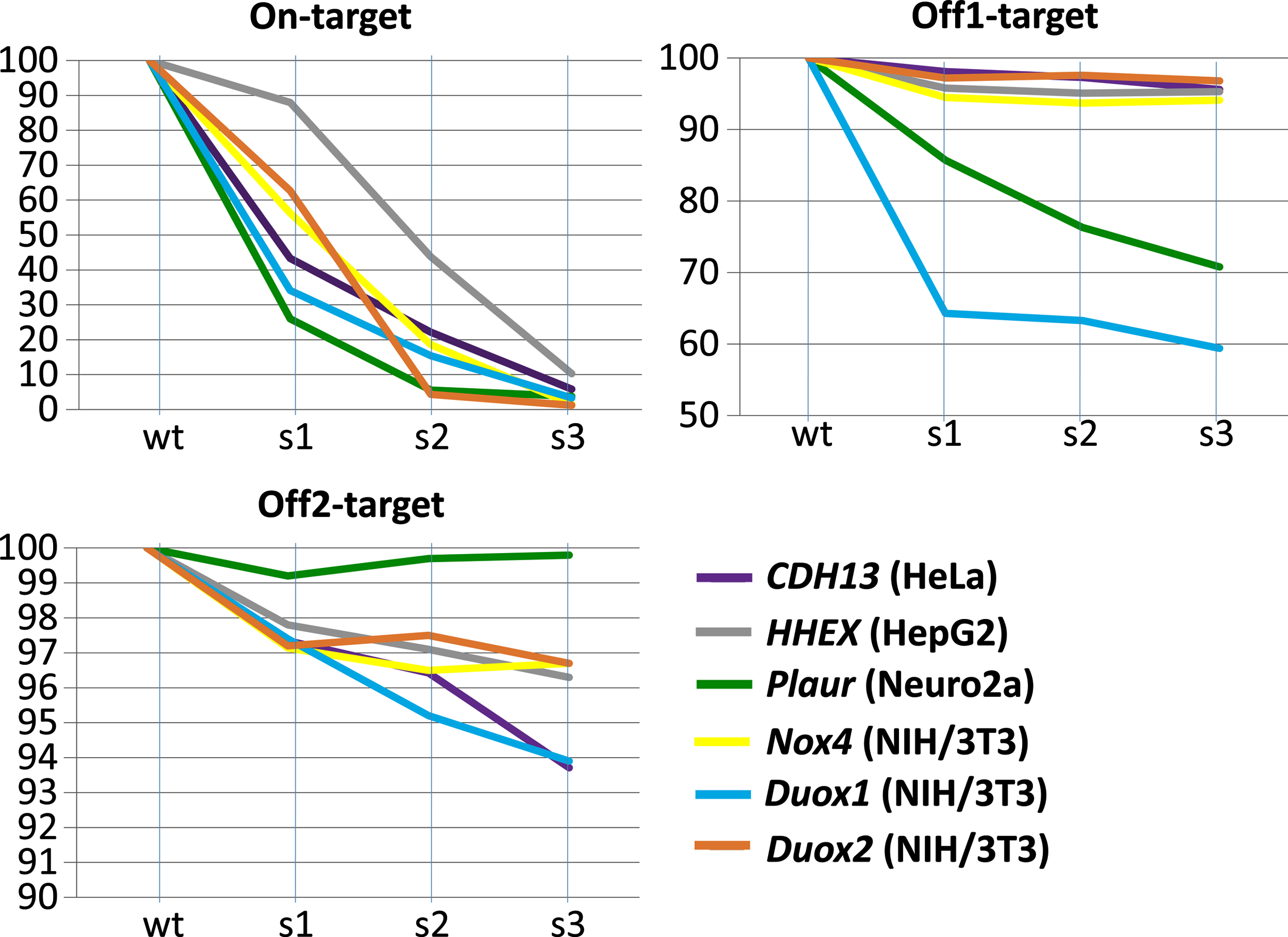
The summary of Sanger sequencing data of on-target and two most likely off-target DNA sites of the cell populations after the first, second, and third rounds of modification. Vertical axis (0Y) corresponding to the percentage of intact On-/Off1-/Off2-target DNA sequences are shown, respectively. s1, s2, and s3, populations after the first, second, and third rounds of modification. Color images are available online.
Discussion
Cell cultures are feasible models to elucidate the role of particular proteins and the mechanisms of disease onset and progression. Their potential and importance increased greatly with the development of simple and efficient genome-editing technologies allowing to alter precisely expression of genes. However, most of routinely used cell cultures, passed numerous passages, are transformed and aneuploid. For example, HeLa cells were obtained more than 65 years ago and their karyotype consists of 70–164 chromosomes—“100% aneuploidy in 1385 cells examined”. 7 The same thing goes with NIH/3T3, HEK, and Neuro2a cell lines.3–5 In such cell cultures, heterogeneity can be observed without any special equipment. Progenies of single-cell clones of NIH/3T3 and Neuro2a cultures growing under the same culture conditions behave diversely: some form plaques or mesh-like structures, others form solid monolayer or huge widely spread cells. We suppose that similar results can be observed with other cell cultures harboring aneuploid genome. This is the consequence of karyotype heterogeneity due to misregulation of mitosis in such cells. Previously, we tried to obtain CDH13-negative HeLa clones using the approach for genome editing of diploid cell cultures, but failed due to named “clonal effect” and restricted efficiency of CRISPR/Cas9 system (from 30% to 60% per allele) 16 (Supplementary Table S6). This forced us to develop the multiround genome modification protocol of general aneuploid cell population, rather than creating single-cell clones.
As we expected, the level of expression of the target proteins in cell populations decreases dramatically with each modification round, but not with similar efficiency. In this study and further, we suppose that all observing efficiency variations depend on the accessibility and transcriptional activity of the target DNA site, gRNA sequence, Cas9 type being used (wild type or nickase), and type and state of the modified cells.17,18
Expectedly, we observed the increasing impairment of target alleles from round to round of modification. In several cases, off-target DNA sites were edited during the first modification round, but off-target activity of CRISPR/Cas9 system was less than on-target (Fig. 5). However, the most unexpected result was obtained when we analyzed sequencing data of potential off-target DNA sites across all modification rounds. In all the cases, we observed that cleavage of potential off-target DNA sites seems to happen very rare in second and third rounds if they have not been cleaved during the first modification round (Fig. 5). Therefore, we did not observe that repeated CRISPR/Cas9 system usage increases the probability of cleavage of a new off-target DNA site, as one could expect. Our data disprove the argument that “CRISPR/Cas9 system promiscuity increases, while being used repeatedly” and makes the offered approach suitable for creating cell models.
Some off-target DNA sites can be predicted using bioinformatics tools, but far from all. 19 During single-cell cloning, the whole progeny inherits the undesired modification that happened in the ancestor clone. These unpredicted and unexpected off-target DNA modifications may influence the observed cell phenotype, and potential interference of undesired mutations with cell aneuploidy may lead to misinterpretation of the obtained results. However, when instead of single-cell clones, we use pooled population of cells with edited genomes, possible off-target effects are usually “diluted” and on-target effects prevail, as on-target activity of CRISPR/Cas9 system in most cases prevails its off-target activity and because the degree of impairment of off-target sites between different clones varies (up to off-target site integrity) (Supplementary Table S2–S5).
In 39.1% of analyzed alleles (9 out of 23), from clones derived from “Hela_s3” population, we observed the identical modification: at least one CDH13 allele of clones #3, 5, 13, 18, 21, 24, 26, and 27 was modified by insertion of one thymidine nucleotide. These support previous observations that nonhomologous end joining mechanism does not repair DNA stochastically, but uses some not well-understood mechanism (perhaps, recognition of microhomology motifs).20,21 At the same time, no protein expression was observed in clones that restored the reading frame. It was reproducible (Fig. 4 and Supplementary Figs. S8 and S9) and definitive answer for this question requires an additional study. We suppose it can be related to lower protein concentration in cell lysates accompanied by insufficient WB sensitivity or to CDH13 protein misfolding or instability due to deletion of six or nine nucleotides, despite reading frame restoration. “Scarless” nucleotide sequence restoration was not observed even in a single allele; otherwise, we would interpret it as an intact (not modified) allele.
In this study, we demonstrated that our protocol can be used for reliable suppressing of expression of various target proteins (enzyme, receptor, and transcription), irrespective of their cellular localization. The only notable shortcoming of the offered approach is its duration (at least 3 weeks). However, it can be shortened by using Cas9- and gRNA-encoding genetic constructs with various selective markers (fluorescent proteins and antibiotic resistance genes), so there will be no need to passage the modified cells until GFP signal fades. An alternative approach for aneuploid cell cultures is to form an artificial population by mixing genome-edited characterized single-cell clones, but we think it is more time- and labor-consuming and unjustified for the most tasks.
The offered approach for aneuploid cell culture genome editing was used in our laboratory for producing several cell populations with knocked-out target proteins. The examples of successful implementation of obtained cell populations for studying of cell physiology can be found in our previous publications.22–24
Thus, we conclude that convenient approach used for creating gene knockouts in diploid cells cannot be applied for gene knockout in aneuploid cell cultures due to their karyotype and phenotype heterogeneity. We offered an approach of repeated rounds of genome editing followed by cell sorting for genome modification of aneuploid cell lines and cells with unstable genome. We showed that with each round of modification, the level of target protein decreases and the degree of editing of the on-target DNA sequence increases. At the very same time, multiroundness of the protocol does not increase the probability of a new off-target DNA site cleavage: it is cleaved after the first round of modification, or is not cleaved at all.
The offered approach as is or in combination with other delivery techniques of CRISPR/Cas9 components can be used for genome editing of aneuploid cell cultures, of cells with restricted proliferation potential, or cells that cannot be single-cell cloned (e.g., HepG2).
Footnotes
Acknowledgments
Study was partially supported by Russian fund for basic research grants #18-315-20053 (obtaining and analysis of HeLa and NIH/3T3 cell lines with knocked-out CDH13 and Duox1/Duox2/Nox4 genes, respectively) and #19-015-00530 (obtaining and analysis of HepG2 and Neuro2a cell lines with knocked-out HHEX and Plaur genes, respectively). Study was conducted using biomaterial collected within the frame of Moscow University Project “Noah's Ark” and equipment purchased as a part of Moscow State University Program of Development.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.