
Abstract
The ideal blood-contacting surface would support endothelial cell lining and suppress platelet adhesion, but, in synthetic biomaterials, these issues often conflict with each other. The reconciliation of this dichotomy may arise by modifying the biomaterial surfaces with “smart” peptides. Phage display is a powerful method for discovering unique peptides capable of binding to target molecules, but the selection of peptides binding to intact cells is an intricate process. In fact, the target molecules are often hindered by the extremely complex composition of cell membrane.
In this work, the traditional phage display screening approach against endothelial progenitor cells (EPCs) was implemented with the introduction of (1) a negative selection step against platelets and (2) the target affinity scoring function phage binding index. The peptide candidates were used to modify an expanded polytetrafluoroethylene (ePTFE) surface to demonstrate that one of them not only has high affinity for EPCs but also simultaneously decreases thrombus formation.
Impact Statement
We presented not only a high-throughput phage display selection protocol for the identification of cell-binding peptides but also a new set of peptides with high affinity to endothelial progenitor cells (EPCs) and with the ability to reduce platelet activation.
The aim of the presented protocol is to decrease the time and cost to identify highly specific peptides against intact cells and/or tissues.
In addition, we proved that expanded polytetrafluoroethylene (ePTFE) modified with the SFKIPYHYDSGQ sequence showed higher EPC proliferation and lower thrombogenicity than naked ePTFE and ePTFE-arginine-glutamic acid-aspartic acid-valine (REDV) peptide.
Introduction
Since 1985,
Peptides, once identified, are easily obtained with a high yield, low cost, and consistent reproducibility.3,4 However, the major challenge of this technology is defining the proper in vitro selection protocol, a process known as biopanning. In biopanning, phage clones that are specifically bound to the target are selected and eluted, while the unspecific or weak binders are removed during the washing steps. 6 The eluted phage clones are amplified in Escherichia coli to enrich the selected phages within the phage library. Subsequently, individual clones are isolated and the primary structure of the binding peptides is deduced by sequencing.
Normally, after several rounds, the library diversity narrows and can converge to a few sequences. However, since the selection process is driven not only by the affinity to the target but also by the phage amplification rate in E. coli phage-growth bias can restrict the selection of target-binding phage to a small subgroup of fast-growing phage clones. Matochko et al., 10 analyzing the enrichment of the sequences, demonstrated that “unreasonable” designed bio-pannings against multisite targets (i.e., cells and tissues) decrease library diversity and limit the number of binding clones that can be identified.
As a matter of fact, the “slow growing” phage clones disappear from the phage library and the “parasite phage” clones (phage with advantage in growth rate) are propagated. These parasitic phage clones exist in the naive libraries but they become “visible” to small-scale sequencing only on serial reamplification of the library. 10 The reduction of amplification steps can minimize this bias in screening against a single target, 11 but it is insufficient in screening against tissue or cells. 12 Therefore, conventional cell-based biopanning approaches13,14 suffer from this limitation, and can often yield ambiguous results and phages with little actual affinity toward the target.
Optimizations, which aim to reduce nonspecific binding and to increase the enrichment of desirable binders, are still being refined. 15 However, there are many difficulties associated with whole-cell screening, including low receptor density and nonspecific adsorption of phage particles. Therefore, a successful cell-based panning is highly dependent on the target of interest, and the methodology needs to be optimized on a case-by-case basis. 15 In several studies, unique binders have been isolated after panning phage libraries against cells either in adherent monolayers or in suspension. 15 Each system has its advantages and disadvantages, and they can be used in case of unavailability of the target in pure form.15,16 Nevertheless, in screening against multisite targets, it is critical to deplete the library of binders to nonrelevant surface targets.15,17 In fact, it is not possible to distinguish between phage clones that bind to the target and those that are connected with “environmental” targets, like the plastic or glass surfaces of labware. 11 The analyses of the sequence diversities by Illumina deep sequencing or enzyme-linked immunosorbent assay are necessary to address this problem. 6
The ability to distinguish true binders from false positives is an important step toward greater integrity of phage display selection. To achieve this, peptides should be compared to known target-unrelated motifs, tested for background binding, and assessed for possible propagation advantages. 12 Further improvements are expected to be achieved in the coming years, although there has been limited research to optimize the cell-based panning. 15 Since there is currently no standardized biopanning protocol to enrich the population of multisite binders in whole-cell biopanning, we previously presented a new ameliorate method. 17 In this article, the method has been improved by introducing consensus sequence screening against peptide/protein databases and compared to the conventional biopanning to provide evidence of its validity (Fig. 1).
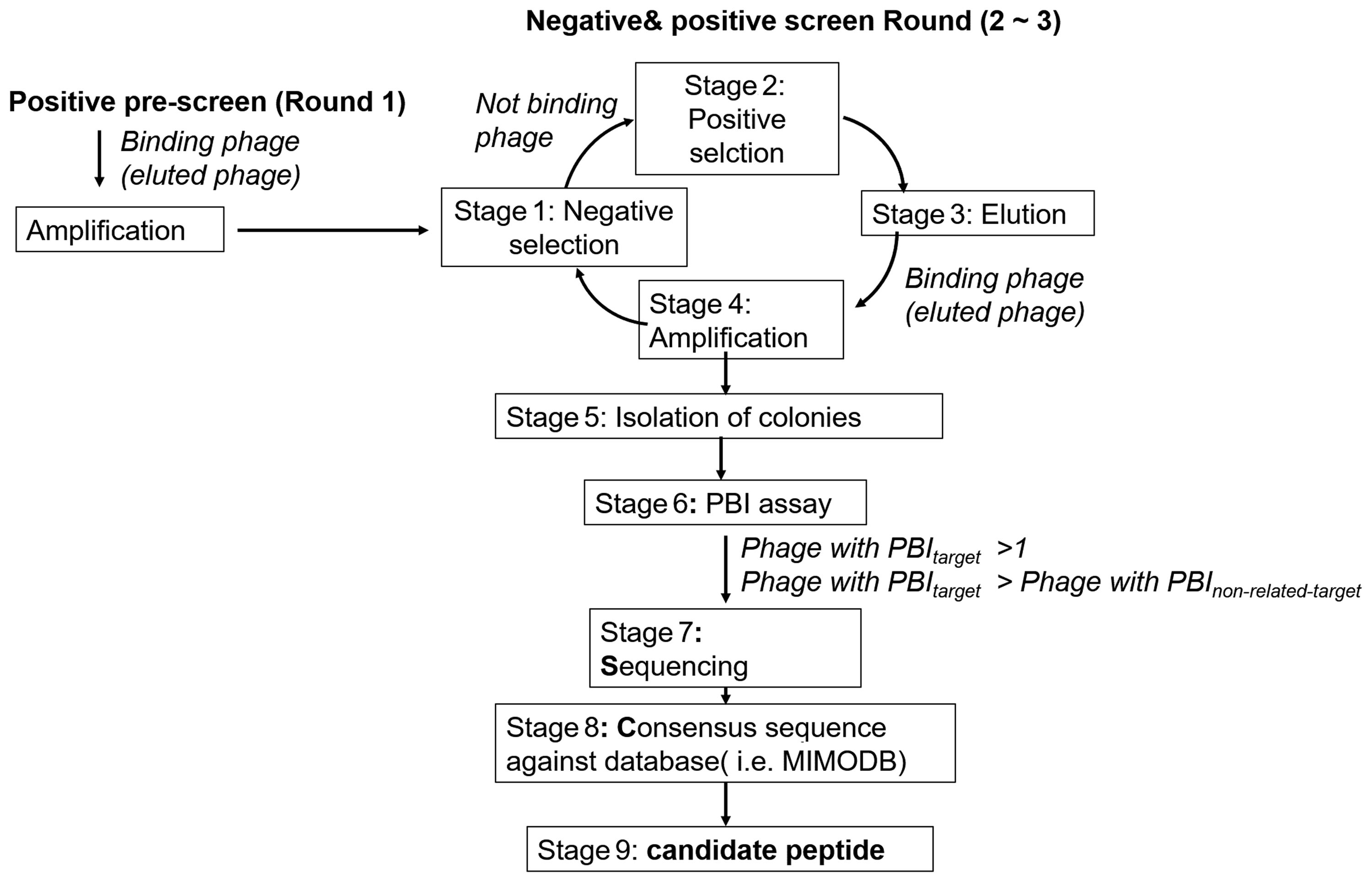
Schematic approach to find candidate peptide.
Materials and Methods
Phage library, bacteria, and cells
The linear heptapeptide (Ph.D.-7) and dodecapeptide (Ph.D.-12) phage display library kits (New England Biolabs, Beverly, MA) were used for the in vitro biopanning experiments, as previously described.17,18 The sequences of the peptides obtained are shown in Table 1. Endothelial progenitor cells (EPCs) (Cat. no. Z7030001) were obtained from BioChain Institute, Inc. (Newark, CA), and cultured in Cell Growth Media Kits EGM-2 SingleQuot Kit Suppl. & Growth Factors (Cat. no. CC-4147; LONZA) at 37°C and 5% CO2. Human hepatocarcinoma cells (HepG2) were obtained from Riken Cell Bank (Tsukuba, Japan) and cultured in Eagle's minimum essential medium (Cat. no. 11995; Gibco-Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum and antibiotics/antimycotics (50 U/mL and 50 mg/mL, respectively; Gibco-Invitrogen), purified single-stranded M13mp18 DNA (Cat. no. N4040; BioLabs).
Peptide Sequences, Type of Biopanning and Phage Binding Index Values
EPC, endothelial progenitor cell; PBI, phage binding index.
Phage binding index assay
Single phage clones (designed as phage-cloneN, where “N” denotes the clone identification number) were used for the phage binding index (PBI) calculation as previously described. 17 In brief, 2 × 10 11 pfu of phage-cloneN was incubated with adherent cells for 1 h at 37°C and 5% CO2. The bounded phages were eluted with 0.2 M glycine-HCl, and this solution was titrated to enumerate the concentration of plaque-forming units, and this was defined as Output titerphage-cloneN. Phage titration of phage-cloneN solution added to the adherent cells was performed, and this was defined as Input titerphage-cloneN. Wild type phage M13 was used as control. The error values were calculated according to the error propagation theory.
Thus, we defined the PBI as follows:

Cell culture on modified expanded polytetrafluoroethylene
GORE PRECLUDE® Pericardial Membrane (expanded polytetrafluoroethylene [ePTFE], Cat. no. PSM-01200) was purchased from Gore Creative Technologies Worldwide (W. L. Gore & Associates, Inc., Newark, DE). N-ɛ-maleimidocaproic acid (ECMA) was purchased from Funakoshi, 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC), 2-(N-morpholino) ethanesulfonic acid (MES), borane solution (BH3 in 1M THF), and the other chemicals were purchased from Sigma-Aldrich (St. Louis, MO). Synthesized peptides were obtained from SCRUM, Inc. (Tokyo, Japan). The samples are indicated as ePTFE-X, where “X” denotes the peptide abbreviation. ePTFE discs (diameter 12 mm) were cut and prepared as previously described17,18 (Supplementary Fig. S1). The samples were stored under vacuum. Surface characterization were performed using ESCA 3400 (Shimadzu Group Company, Kyoto, Japan) and static water contact angles were detected using the contact angle and surface tension meter CA-X (Kyowa Interface Science, Saitama, Japan) (Supplementary Table ST1). Three measurements on different surfaces were taken for each sample.
The peptide-immobilized ePTFE discs were rinsed with sterile water and then placed in 24-well plates as previously described.
17
The discs were set on the bottom of the each well under a stainless-steel ring, 2 × 10
4
cells (EPCs) were seeded and cultured for 4 or 24 h. Experiments were conducted in triplicate. Cell counting was conducted using Cell Counting Kit-8 (Dojindo, Kumamoto, Japan), and the results were recorded on a Varioskan microplate reader (ThermoFisher, Yokohama, Japan). The percent of cell growth (CGI, cell growth index) was expressed by the following equation:
Thrombogenicity assessment: whole blood clotting
The quantification of blood coagulation was conducted by adapting the Harboe spectrophotometric assay for the measurement of hemoglobin (Hb).19–23
Whole blood was collected from pigs using Anticoagulant Citrate Dextrose A anticoagulant. The whole blood samples were recalcified with the addition of 0.1 M CaCl2. The modified ePTFE samples were rinsed with sterile water and placed in 24-well plates. The discs were set on the bottom of each well using a stainless-steel ring. The modified ePTFE, nonmodified ePTFE, and empty wells of the polystyrene (PS) plate (negative control) were incubated for 5, 10, and 15 min at 37°C with 15 μL of recalcified blood. The red blood cells not trapped in the thrombus were lysed by adding 300 μL of distilled water and incubating for 10 min at room temperature.
20
All of the samples were appropriately diluted, and the concentration of released Hb was measured by transferring the supernatant to a 96-well plate followed by spectrophotometric analysis. The absorbance was read at 380, 415, and 450 nm against a distilled water blank in an Varioskan microplate reader, according the method devised by Harboe.21–23
The concentration of Hb was calculated using the following equation:
The terms A415, A380, and A450 are the absorbance of hemolyzed samples at 415, 380, and 450 nm, respectively, all within the linear range of the spectrophotometer (0–2 absorbance units). These results were compared to the solution of distilled water with not-calcined whole blood sample to define the blood clotting index (BCI) as follows:
where HbN is the Hb in each sample. Therefore, higher value of BCI corresponds to less thrombogenicity in the samples. The error values were calculated according to the error propagation theory. 24
Samples used for scanning electron microscopy (SEM) were fixed in formaldehyde (3.7% v/v in phosphate-buffered saline) for 10 min and dehydrated with graded ethanol (50%, 60%, 70%, 80%, 90%, and 100%). The samples were then dried under vacuum, coated with gold by sputtering (EIKO IB3 Ion coater; EIKO Engineering CO., Ltd., Japan), and then subjected to SEM observation (JCM 5700 JEOL, Japan).
Results and Discussion
Peptides selection
A phage display peptide library contains 10 9 –10 10 different clones. A vast majority of clones do not bind to the target molecule, a small proportion of phages bind with low affinity to the target, and only a few of them are capable of high-affinity binding. Therefore, in typical biopanning, the clones that do not bind are removed during washing steps, and the phages with low and high affinity are eluted and subjected to additional rounds of selection. Several rounds are necessary to obtain phages with a high affinity for the target, and these steps can cause the loss of promising phage clones.10,11 In fact, phages can bind to other components of the screening system rather than the target, 12 and certain phages can propagate faster in host bacteria. 12 Decreasing the number of amplification steps can reduce the selection bias imparted by differences in growth rate. However, a reduced number of rounds may not be sufficient to individuate the most effective phages, especially with complex, multisite targets, such as the cell membrane. Therefore, we modified the original biopanning protocol by adding a negative selection step to improve the selection stringency. 17 In fact, it is possible to influence the selection by performing some negative selection steps before positive selections to remove unwanted specificities, but the high risk of losing the best binders is the limiting factor in this procedure. However, applying negative selection to initial library is not considered advantageous. Thus, after an initial positive selection step and amplification of these eluted phage-clones, the input for the next biopanning round will be made by phage clones that have “survived,” so they are the best binder for EPCs. Consequently, performing the negative selection prior the next positive selection step can effectively remove target-unrelated phage clones and retain the relevant phage clones (best binders). However, when negative selection is used to reduce unwanted selection, the amplification in bacteria after this step should be avoided. In fact, some phage clones may have a growth advantage in bacteria, no affinity to the unwanted target, and still have low affinity to the target. Amplification steps will increase the number of such undesired phage clones, increasing their likelihood of being selected as potentially helpful. Therefore, the positive selection step was performed just after the negative selection, and the eluted phage clones were amplified only after these steps were completed. In addition, we used the PBI scoring function to experimentally determine the affinity of each phage clone to the desired target (EPCs). In the PBI assay (Table 1), a wild type M13 phage was used as a “control” phage. When the M13 and phage-cloneN bind with similar affinities to the target, the PBI ratio is close to or less than 1. Instead, PBI ratio higher than 1 is connected with an increase of affinity to target. However, the enrichment of sequences is driven by affinity to both target-related (i.e., EPCs) and nontarget-related (i.e., Petri dish or a common class of cell membrane proteins), thus, we implemented this procedure comparing the PBI obtained against EPCs to that obtained against HEPG2. In fact, phage clones which bind similarly to EPCs and HEPG2, or bind to noncell-related target (i.e., plastic, collagen) are characterized by similar PBI (PBIEPCs = PBIHEPG2). Instead, a higher PBIEPCs value indicates that the phage clone is highly specific for EPCs. 17 In this study, we used HEPG2 cells, but any other cell type could have been used, as negative control. In fact, EPCs and HEPG2 are different cell lines with a variety of different proteins and receptors on the cellular membrane, thus phage clones should exhibit a radically different PBI value (PBIEPCs ≠ PBIHEPG2) (Fig. 2A, B).
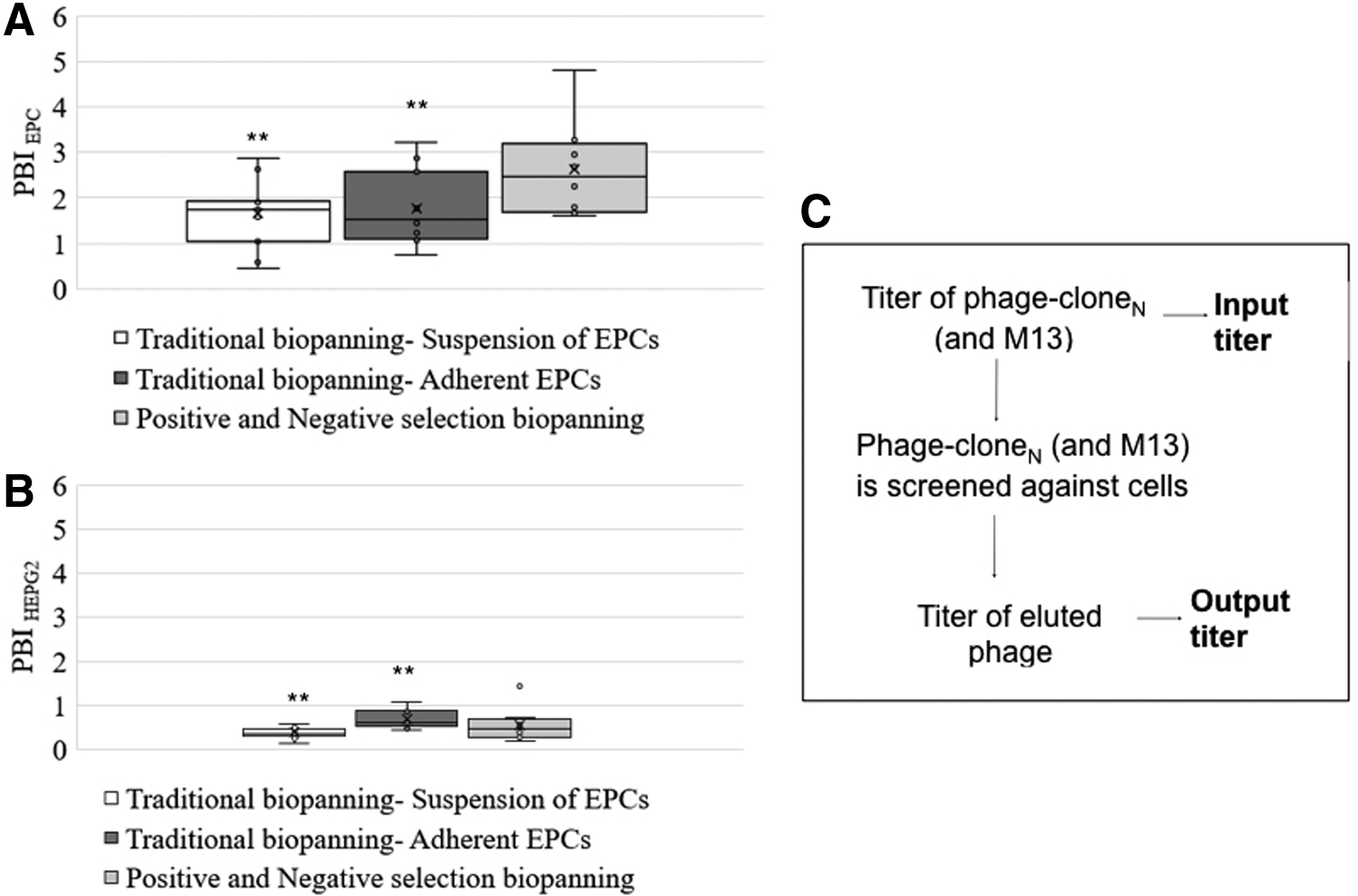
Box plot graph shows the PBI results:
The PBIEPCs variation in the phage clones selected by traditional and modified biopanning was compared using a box plot graph (Fig. 2A and Table 1). The PBIEPCs in the traditional biopanning was smaller than the modified Negative-Positive biopanning. This is a clear evidence of the higher stringency of the second selection method. The low level of PBIEPCs obtained from traditional biopanning against a suspension of cells is probably due to the effect of trypsinization in preparing the cell suspension. Trypsin treatment cleaves plasma membrane proteins such as fibronectin-specific receptors, VCAM-1, and E-selectin, 25 and the surface property of the trypsinized cells may not retain their native states. The folding, quaternary structure, expression level, association with neighboring proteins, and the biological functions of proteins may be changed. In addition, cell suspension contains not only the fully intact healthy cells but also dead cells, cell debris, and cells that do not express the targeted protein on their surfaces, which can often lead to isolation of nonspecific binders. 15 Interestingly, no significant PBIEPCs difference was detected comparing the peptides selected against the suspended or adherent EPCs. However, a clear difference is visible in the set of results from HEPG2. These results are unexpected, probably the trypsin is going to damage not all the proteins and receptor in cell membrane. Whereas, the PBIHEPG2 values (Fig. 2B) are about 1 or smaller: this indicated that these sequences do not have high selectivity for HEPG2.
Subsequently, the phage clones with (1) PBIEPCs higher than 1 and (2) PBIEPCs higher than PBIHEPG2, were sequenced and, the obtained peptides were analyzed in the peptide database MimoDB.26,27 We found that sequences A, B, and L (Table 1) are in the MimoDB database, but none of these peptides bears any target unrelated peptide motif (Supplementary Fig. S2). Therefore, they may be multiple target “binders,” not “noise” (i.e., plastic, Petri dish). In Table 1, the peptide sequences selected in this article are reported.
Cell culture on modified ePTFE
The X-ray photoelectron spectroscopy and contact angle (Supplementary Table ST1) characterization proved that the all samples were properly functionalized by the different peptides selected by phage display. In these experiments, naked ePTFE and ePTFE modified with arginine-glutamic acid-aspartic acid-valine (REDV) peptide are considered as negative and positive controls, respectively. In fact, when REDV, adhesion ligand peptides, is immobilized on cell, no adhesive substrates, endothelial cells attached and spread, and it has been shown that endothelial cell monolayers on REDV-grafted substrates are nonthrombogenic and improve the patency in vivo.28–30
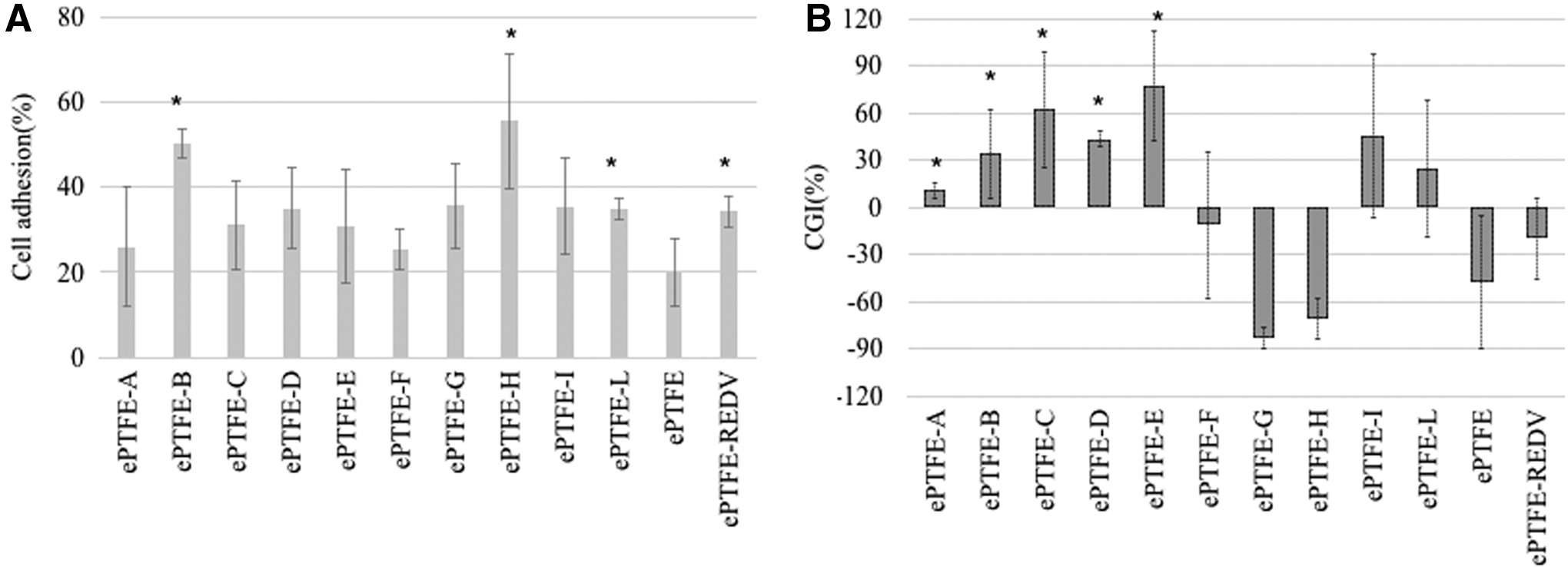
Figure 3A shows the percentage of adherent EPCs. After 4 h, ePTFE-B, ePTFE-H, and ePTFE-REDV greatly enhanced the cell adhesion in comparison to naked ePTFE. In particular, more than 50% of the seeded cells attached to the ePTFE-B and ePTFE-H surfaces. However, after 24 h, samples ePTFE-F, ePTFE-G, ePTFE-H, and ePTFE-REDV exhibited a negative CGI, indicating a cell detachment. Cell growth (Figure 3B) was detected in ePTFE-C, ePTFE-D, ePTFE-E, ePTFE-I, and ePTFE-L. Three of these peptides were selected by Negative-Positive biopanning (C, I, and L), and two using the traditional method against adherent cells (D and E). As expected, the peptides selected only by the selection against EPCs (F) in suspension showed low cell adhesion and very high cell detachment.
Thrombogenicity assessment: whole blood clotting
Whole blood was used to determine the clotting time as a measure of thrombogenicity. Citrated plasma serves as control, as it does not cause clotting, and a naked PS well was used as a negative control. Using the equation of Harboe21–23 the free Hb in the solution was determined and connected with the clot formation. When clotting occurs, red blood cells are retained in the clot, and less Hb is released by lysis upon the addition of distilled water. Results, expressed as BCI, show (Fig. 4A) that all the samples (except ePTFE-F and ePTFE-H) are less thrombogenic than naked ePTFE. It is well known that hydrophilic surfaces can improve biocompatibility, inhibiting platelet adhesion and activation. These results show that although ePTFE-C and ePTFE-I have hydrophilicity similar to naked ePTFE, they have much less thrombogenic surface. It is interesting to note that the peptides selected by Negative-Positive biopanning have not only provided a surface with lower thrombogenicity, but also improve EPC growth. Peptides A, B, and L, found in MimoDB, stimulated EPC proliferation slightly. Peptides A, B, and H, that were selected by two different methods (adherent and suspended EPCs), showed a lower PBIEPCs (Fig. 4B). These results suggest that peptides A, B, and L are probably multitarget-binder peptides. In addition, these results have further strengthened our hypothesis that PBI is a useful index to evaluate the quality of the selected peptides. In fact, samples A, B, H, and L peptides have lower PBIEPCs (Fig. 4B and Table 1).
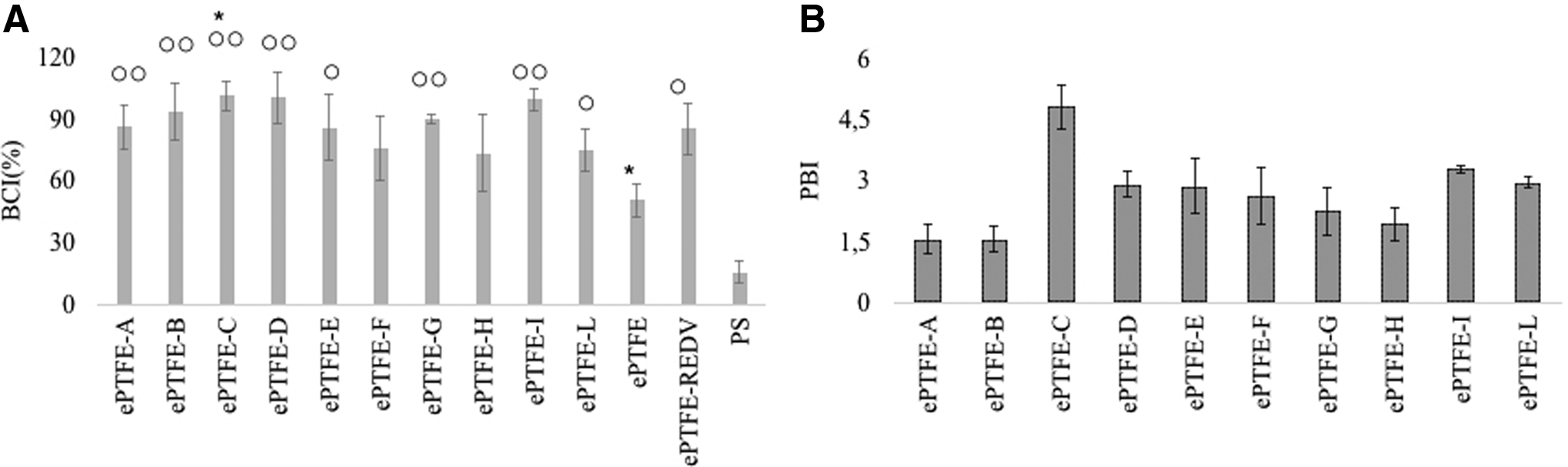
Figure 4A and Supplementary Figure S3 show SEM pictures of the sample surface after contact with calcined whole blood for 10 min. On the surface of ePTFE-B, ePTFE-C, ePTFE-D, and ePTFE-I, fibrin deposits and alpha granules were observed. The surfaces of the other samples contained several erythrocytes and platelets. Figure 5B and Supplementary Figure S3 show the dynamic blood clotting profiles. The calcined whole blood on PS (negative control) completely clotted after 10 min, while ePTFE showed a rapid BCI decrease, and almost half of the blood clotted within 10 min. However, during the washing steps for SEM sample preparation, the clots were easily removed from ePTFE surface. This did not happen on the other samples. This observation appears to indicate that clots from an ePTFE surface detach and might be transported in the blood stream.

Comparison of representative SEM images and BCI. ePTFE-C, naked ePTFE, and PS are compared as representative samples.
When blood is in contact with a foreign surface, plasma proteins are adsorbed and then platelet adhesion and the coagulation process start. Blood compatibility is reached when there is not too much interaction of platelet with the material surface. Therefore, our studies show that ePTFE-C, ePTFE-B, ePTFE-D, and ePTFE-I can increase the clotting time of naked ePTFE better than ePTFE-REDV.
Our results demonstrate that Negative-Positive biopanning not only improves the stringency of selection and decreases the amplification rate bias but also helps to remove unwanted binders from the phage library. In addition, this method can be useful in gene delivery systems based on highly specific recognition of tissue surface proteins, and for the specific targeting of organs. Moreover, it can be used to select peptides to improve the design of prosthesis and devices. In fact, it is possible to perform negative selection against bacteria, in particular, against biofilm bacteria associated with prosthetic infections. Thus, the use of negative and positive selection method to identify peptides might reduce the infections in orthopedics specifically, and surgery in general, with major consequences for the patient and health care system.
In this work, a set of peptides with high affinity to viable EPCs and low interaction with platelets was identified in only three rounds of selection. In addition, we proved that peptide-modified ePTFE surface improves the EPC adhesion and decreases platelet activation. In particular, one of these peptides (SFKIPYHYDSGQ) improved EPC growth and decreased the thrombogenicity of ePTFE surface better than the REDV sequence.
Conclusion
In this article, we introduced not only an improved protocol for the identification of cell-binding peptides but also a new set of peptides with high affinity to EPCs and the ability to reduce platelet activation. The aim of the presented protocol was to decrease the time and cost to identify high affinity peptides against intact cells and tissues. The key components of this approach are the use of (1) a double screening procedure (Negative-Positive biopanning) and (2) a novel analytical tool, PBI: a fast and inexpensive target-affinity assessment. In addition, we proved that ePTFE modified with the SFKIPYHYDSGQ sequence showed higher EPC proliferation and lower thrombogenicity than naked ePTFE and ePTFE-REDV.
Footnotes
Acknowledgment
The authors gratefully acknowledge the help and assistance with blood sampling of Atsushi Mahara. This work was supported by the S-innovation Research Program for the “Development of the biofunctional materials for realization of innovative medicine,” Japan Agency for Medical Research and Development (AMED).
Authors' Contributions
M.C.M.: conception and design, data collection, data analyses and interpretation, and article writing. T.Y.: conception and design, data interpretation, and article correction.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.