
Abstract
Translational studies to elucidate the response of immature bone to biologic and physical stimuli have been held back by the lack of a viable long-term functional bone explant model. This study attempts to bridge this gap between cell culture and animal model studies. In this study, we describe a methodology to derive a 300 μm organotypic femur slice comprising physiological zones (epiphysis and meta-diaphysis) essential for endochondral bone development. The unique capability of slice culture model incorporating enhanced nutrient access to distinct bone tissue components associated with linear bone growth facilitates the investigation of the orchestrated cellular transition of chondrogenic and osteogenic cells involved in endochondral bone development in an ex vivo setup. Bone slices of 300 μm were prepared from 4-day-old postnatal rats and were viable in culture up to 21 days. On days 7 and 15, an increase in chondrogenic and osteogenic modulations was confirmed in epiphysis, metaphysis, and diaphysis. An increase in osteocytes, osteoblasts, and hypertrophic cells were found at these time points, as well as a noticeable increased expression of chondrogenic and osteogenic markers (collagen II, Runx2, and osteocalcin) confirmed endochondral progression. Osteoclast-mediated bone resorption was demonstrated on day 15 by tartrate-resistant acid phosphatase staining. Attenuated total reflection infrared spectroscopic analyses, furthermore, confirmed a time-dependent increase in phosphate levels, bone minerals, and hydroxyapatite for 15 days. Our establishment of a bone slice culture model closely mimicking the in vivo cellular transitions and endochondral microenvironment of a mineralizing bone provides a vital new tool for the elucidation of cellular and endochondral mechanisms of bone development, maturation, and growth plate modulations. The presented model has the potential to be utilized in implementation of preclinical, toxicological, and therapeutic investigations.
Impact Statement
So far, in vivo studies are applied as model organism and are considered as “gold standards” to obtain reliable results for clinical applications. However, conventional in vivo model systems are largely associated with high maintenance costs and strict ethical legislations. This study focuses to establish an ex vivo bone model that permits the screening, evaluation, and prediction of bone tissue responses to cell transplantation, biomaterials, and biological and pharmacological substitutes, thereby providing a potential platform to test novel therapeutic strategies to restore, repair, and manipulate developing bone in situ.
Introduction
Three-dimensional (3D) organotypic cultures—organ/tissue slices are anticipated to bridge the gap between 2D in vitro cell culture and in vivo experimental systems. Ready access to different cell types, the possibility to perform real-time imaging of cellular development and physiological modulations, and localized addition of mediators or pharmaceutical agents make slice cultures ideal ex vivo models. 1 Organotypic slice cultures critically provide effective distribution of nutrients and metabolites for correspondingly longer ex vivo tissue survival than whole organ cultures, which were popular models in the past.2–5 Organotypic slice cultures recapitulate aspects of the structural and connective organization of a tissue or organ of interest to varying degrees, allowing the analysis of complex cell dynamics such as cell viability, proliferation, migration, and differentiation in a tissue microenvironment. Unlike 2D cultures, they also enable optical access to native stromal tissue and real-time analysis of physiological modulations. 6
Organotypic slice cultures are most widely used for central nervous tissue studies, but have also been established from heart,7–9 lung, 10 liver, 4 intestine, 11 and recently from brain 12 and pancreatic tumor tissue. 13 Bone, being a hard tissue, is relatively challenging to culture in an ex vivo setup due to its complex tissue architecture and calcified nature.14,15 A variety of ex vivo whole bone cultures have been developed for specific applications, namely to study linear bone growth (mouse metatarsal culture), bone and cartilage developmental mechanisms (mouse femoral head culture), or bone responses to mechanical loading (bovine trabecular core culture).16–22
A 2 mm rat mandibular pulp slice culture has been established to study regenerative mechanisms in bone repair,23,24 specifically by injecting dental pulp stem cell into these slices. An ex vivo model enabling detailed analysis of mechanisms underlying longitudinal bone development with access to different developmental zones has, however, not yet been established so far. 17 We hypothesize that bone slice cultures can be cultivated similar to soft tissues. Our aim was, thus, to establish a femoral bone organotypic slice model allowing diffusion of nutrients and extracellular factors of femur consisting of the epiphyseal and meta-diaphyseal zones responsible for endochondral ossification. Applying methods established for organotypic slice models of soft tissue organs,13,25 we precision cut 300–400 μm slices from femurs harvested from 4-day-old postnatal rats, cultured them under osteogenic conditions, and documented the ensuing endochondral cellular transitions.
Materials and Methods
Experimental design
For each experiment, femurs from 4-day-old postnatal rats belonging to the same litter were explanted to derive 300 μm tissue slices. The resultant slices were randomly assigned to different experimental groups and cultured in osteogenic medium. Depending on the analysis, the cultures were harvested on days 3, 7, and 15 and were evaluated for viability and osteogenic properties in comparison with those of noncultured 4-day-old postnatal femurs. Each experiment was repeated at least three times to confirm the results. All histological analyses were confirmed by staining at least three sections per slice. (Detailed description of tissue preparation and analysis can be found in respective Methods sections and Supplementary Materials and Methods section.)
Preparation and culturing of organotypic culture slices
Femurs were aseptically explanted from postnatal day 4 Sprague Dawley rats weighing ∼13 g after sacrifice by decapitation, and washed in ice-cold Dulbecco's phosphate-buffered saline (DPBS; Gibco, Invitrogen, Austria) for 1 min to remove surgical residues and blood. The explants were then embedded by placing them in prechilled stainless steel molds (Tissue-Tek, Sakura, Japan) in molten 2.5% agarose and cooling on ice to accelerate solidification. A VT1200 Leica vibratome calibrated according to manufacturer's instructions was used to prepare 300–400 μm coronal femur sections. The embedded explants were mounted on the specimen plate using tissue culture grade cyanoacrylate glue (Roti® coll1; Carl Roth, GmbH, Austria) and cut with a stainless steel razor blade (Gillette Procter & Gamble, United Kingdom) under buffered conditions using ice-cold 1 × Dulbecco's phosphate buffered saline, specimens at an amplitude of 1.5 m, and a sectioning speed of 0.6 m/s.
The resultant slices were then gently washed for 30 s in α-minimum essential medium (α-MEM; Sigma-Aldrich, St. Louis, MO) and antibiotics penicillin (100 U/mL) and streptomycin (100 μg/mL; Invitrogen, Carlsbad, CA) and transferred into six-well plates containing a Millcell cell culture 30 mm insert with pore size of 0.4 μm (Merck, Darmstadt, Germany). Each well was supplemented with 1 mL of osteogenic medium containing α-MEM, antibiotics,
Live and dead staining
Cell culture viability was determined by live/dead staining using the protocol described in the Supplementary Materials and Methods section.
Tissue processing for histological evaluations
See Supplementary Materials and Methods section.
Histological and immunohistochemical stainings
Hematoxylin and eosin staining
Hematoxylin and eosin (HE) staining was used for general cell identification as previously described. 26 Slices were examined on days 7 and 15 and compared with noncultured control slices (NCCSs).
Movat's pentachrome (Verhoeff) staining
Movat's pentachrome staining (Morphisto Austria) protocol was adapted from a previous study 27 by replacing Weigert's iron hematoxylin solution with Verhoeff's solution provided in the kit. Fifteen-day cultured slices and NCCSs were evaluated for cartilage mineralization (sea green), mineralized bone matrix (pale yellow), native cartilage matrix (bright yellow), and cell nuclei (reddish brown).
Alizarin red staining (calcium turnover)
Deparaffinized and rehydrated sections were stained with alizarin red solution (pH 4.1) for 3 min dehydrated in acetone (Merck) and acetone–xylol solution (1:1, 20 dips per section), cleared in xylol (2 × 3 min; Merck), and mounted with Roti. Cultured slices and NCCS controls were examined on days 7, 3, and 15.
Tartrate-resistant acid phosphatase staining
Fifteen-day cultured slices and NCCS controls were rehydrated and incubated for 20 min at room temperature (RT), 20–25°C, in 0.2 M acetate buffer (pH 4.5) containing 50 mM tartaric acid (Sigma) and 0.2 M sodium acetate. The sections were then stained with 0.2 M acetate buffer, naphthol AS-MX phosphate, and fast red (Sigma) at 37°C, rinsed in distilled water and mounted with Aquatex (Merck).
Immunohistochemistry
Before immunohistochemical staining, sections were deparaffinized in xylene (2 × 10 min), rehydrated in different grades of ethanol (100%, 90%, and 70% for 5 min) and washed in Tris-buffered saline (TBS; Santa Cruz, Germany). Antigen retrieval was performed by incubation in 20 μg/mL proteinase K solution (Abcam, Cambridge, United Kingdom) for 7 min at RT. Endogenous peroxidase was then quenched by incubation in 0.3% hydrogen peroxide (Merck) for 30 min at RT and washing with TBS (2 × 5 min). Nonspecific binding was blocked by incubation in normal goat serum (Dako, Denmark) for 30 min at RT. Sections were incubated with monoclonal mouse anticollagen II (1:200 in PBS) (Clone: II-4C11, AF5710; Acris Antibodies, CA) and monoclonal mouse antiosteocalcin (1:1000 in PBS) (Clone No. OC4-30, LS-C73941; LifeSpan BioSciences, WA) for 60 min at RT.
For Runt-related transcription factor 2 (Runx2) staining, a polyclonal rabbit anti-Runx2 (1:500 in PBS, ab102711; Abcam) was used with overnight incubation at 4°C in a humidified chamber. Sections not incubated with these primary antibodies served to rule out nonspecific binding. After removal of excess primary antibody, sections were washed in TBS (Santa Cruz), distilled water (2 × 5 min), and then incubated with EnVision+ System-HRP polymer conjugated to antimouse or antirabbit secondary antibodies (Dako, CA) for 30 min at RT. Color development was performed using the Liquid DAB+Substrate Chromogen System (Dako) for 20 min at RT followed by washing in distilled water (3 × 2 min). Sections were counterstained with Mayer's hematoxylin solution (Carl Roth) and mounted with Aquatex and subjected to initial microscopic evaluations as described in a previous study 28 and in Tissue Processing for Histological Evaluations section in Supplementary Materials and Methods.
Computerized morphometry
Six fields (each with an area of 600,000 μm2) were randomly selected from each section. Blinded evaluations of osteocalcin, Runx2, collagen II, and alizarin red were performed by two investigators as previously described. 29 Quantification results are presented as mean ± standard deviation.
Attenuated total reflection infrared spectroscopy
Attenuated total reflection infrared (ATR-IR) spectroscopic measurements were performed using a Bruker Alpha ATR-IR spectrometer (Bruker, Germany) equipped with the ALPHAs Platinum ATR single reflection diamond over a scan range of 4000–650 cm−1. A total of 32 scans were performed (using OPUS/mentor software, version 6.5) with a resolution of 4 cm−1. Samples were dried in a stream of liquid nitrogen and scanned at three different spots, that is, epiphysis, metaphysis, and diaphysis.
Results
Organotypic femur slices survive in culture for a minimum of 22 days
Organotypic femoral slice cultures (300 μm) were prepared from 4-day-old rats as shown in the schematic description in Figure 1A. Tissue viability during culture was evaluated by live/dead (red/green) staining on days 4, 7, 15, and 22 (Supplementary Fig. S1). Tissue slices displayed viability up to 22 days, although a noticeable increase in the number of dead cells was evident on day 22. Considering these observations, only slices cultured for up to 15 days were included in the subsequent analyses. Controls showing live and dead bone tissue are presented in Supplementary Figure S2.

Schematic overview of preparation of femoral bone slices (OTCs) and assignment of respective zones in the OTCs for histological evaluations.
Time-dependent morphological changes in femur slice cultures
To facilitate comparable morphological studies, the postnatal femur was empirically divided into distinct zones defined primarily according to the arrangement of specific cell types, respective tissue origin, and differentiation processes. Different approaches have, however, resulted in some variation in these definitions. We refer to the zones as depicted in Figure 1B using terminology based on the study published in 2015 by Akkiraju and Nohe. 30 Time-dependent morphological changes of cultured femur slices were assessed on culture days 7 and 15 by HE staining in comparison with HE-stained NCCSs. The epiphyseal overview revealed (Fig. 2A-i to iii) more intense staining in the superficial zone and transitional zone (TZ) in day 7 cultures compared with controls (NCCSs), which displayed homogeneous staining. This was followed on day 15 by noticeably reduced staining in potential ossification sites (secondary ossification center, zone of calcification).
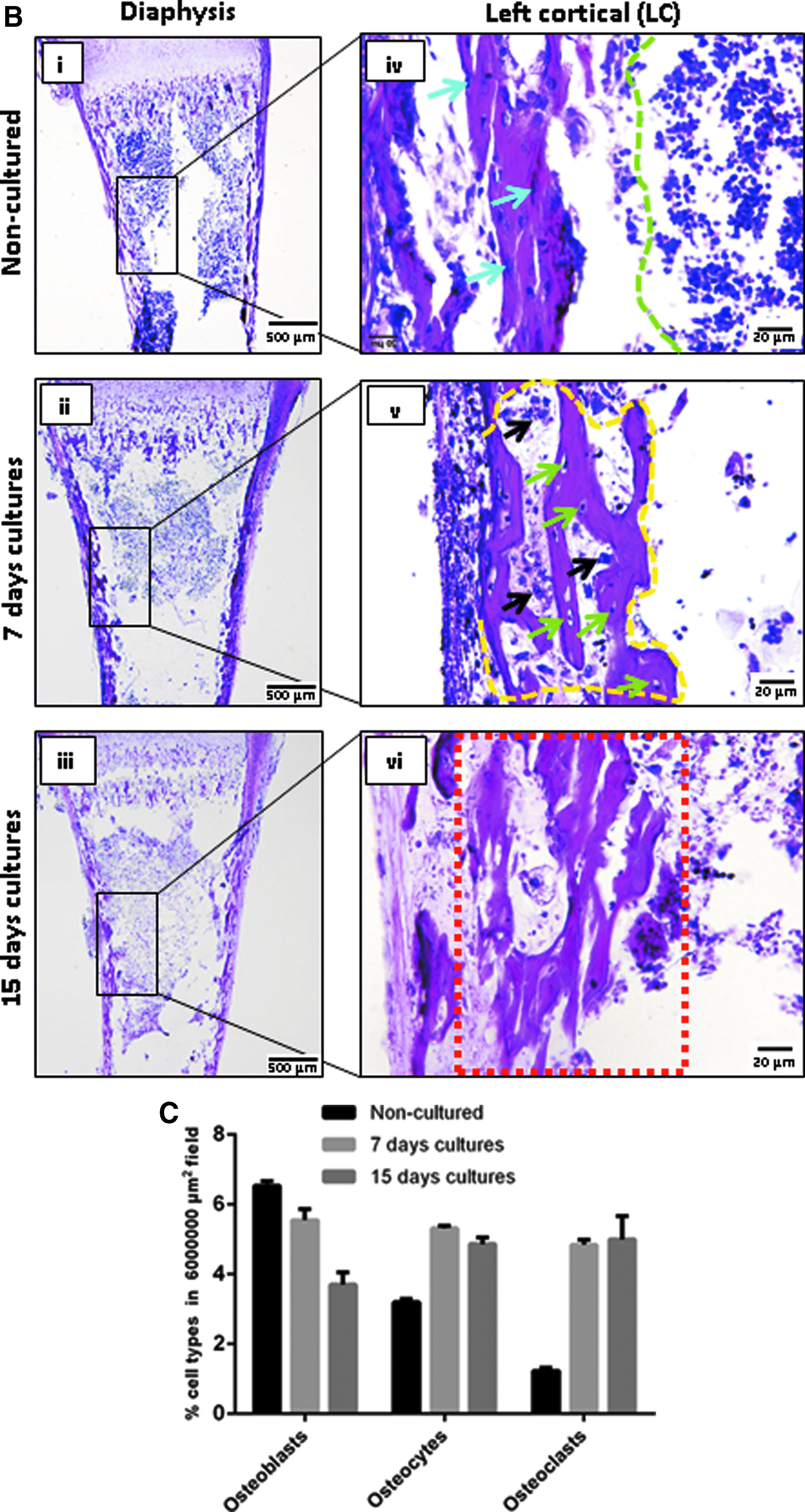
HE stainings demonstrated endochondral morphological transitions in cultured slices compared with noncultured slices (NCCSs).
Higher magnification microscopic evaluations supported this primary indication of time-dependent tissue modulations within the distinct epiphyseal zones. On day 7, in the TZ, quiescent cells detected in NCCSs (Fig. 2A-iv, green arrows heads) were replaced by isogenous cell groups containing several premature chondroblasts (Fig. 2A-v; green circles and yellow arrows, respectively). On day 15 (Fig. 2A-vi), lacunae containing single chondrocytes with enlarged nuclei (green asterisk) were surrounded by interterritorial matrix (black arrows), indicating maturation of cell aggregates into hypertrophic chondrocytes. In the middle zone (MZ), flattened stacks of columnar chondrocytes and insular cell groups (Fig. 2A-vii) were superseded by a scattering of columnar chondrocytes and an increase in chondrocyte-associated lacunae on day 7 (Fig. 2A-viii, yellow circles).
A subsequent increase in scattered single cells with pyknotic nuclei indicating chondrocyte hypertrophy was observed on day 15 (Fig. 2A-ix; yellow asterisk). In the deep zone (DZ), the apparent disintegration of hypertrophic chondrocytes in enlarged lacunae (NCCSs, Fig. 2A-x) observed on day 7 was advanced on day 15 by large elongated and empty lacunae surrounded by densely calcified extracellular matrix, respectively (Fig. 2A-xi, red circle; xii; red arrows), indicating chondrocyte degeneration and mineralization processes in the DZ.
The morphological transition into osteogenic tissue was most advanced in the diaphysis compared to epiphysis (Fig. 2B-i-vi). In the cortical diaphysis, a monolayer of dark-stained thin and elongated cells in the peripheral bone collar (osteoblasts; blue arrows) adjacent to the bone marrow in the medullary cavity (green margin) (Fig. 2B-iv blue arrows) in NCCSs gave rise to trabecular spicules (yellow margins) containing entrapped osteocytes (green arrows) and structures resembling multinuclear osteoclasts (black arrow, Fig. 2B-v) by day 7. On day 15, the bone marrow appeared to be condensed into calcified thickened bone collar (red margin), indicating advanced diaphyseal osteogenic differentiation and mineralization (Fig. 2B-vi).
The results obtained by HE staining were substantiated by histomorphometric quantification of bone cell types in the diaphysis. Number of osteoblasts decreased progressively and substantially during femur slice culture (5.54% ± 0.31% on day 7 and 3.70% ± 0.34% on day 15 vs. 6.53% ± 0.13% compared to NCCSs; Fig. 2C). Number of osteocytes and osteoclasts, in contrast, were distinctly higher than those on 7 days (5.31% ± 0.07% and 4.84% ± 0.13%, respectively) and remained unchanged on day 15 (4.86% ± 0.18% and 4.99% ± 0.66%, respectively) compared to NCCSs (3.19% ± 0.08% and 1.23% ± 0.08%), demonstrating an improved osteogenic lineage in cultured slices. As anticipated, the mentioned changes were more advanced than 19-day NCCSs, although the cellular transitions in cultured slices resembled changes in TZ, DZ, and diaphyseal shaft of 19-day NCCSs (Supplementary Fig. S3).
Chondrogenic and osteogenic modulations in cultured femur slices
Movat's pentachrome staining was used to distinguish mineralized regions from nonmineralized regions in the slice cultures as described in Materials and Methods section (Fig. 3A). The following analyses were only performed on 15-day cultured slices, due to predominant changes detected at this time point by HE staining.

Endochondral differentiation induced mineralization of extracellular matrix and initiated osteoclast-mediated resorption of mineralized bone in both epiphysis and diaphysis.
The fundamental matrix of the epiphysis exhibited green and pronounced yellow staining compared with pale yellow staining for NCCSs, indicative of collagen matrix mineralization during culture (Fig. 3A-i, ii). Further evaluation in specific zones at higher magnification revealed the appearance of large hypertrophic cells embedded in a dense intensely stained territorial matrix in TZ (Fig. 3A-iv, inlet red arrows heads), underlining the maturation and mineralization of the native cartilage matrix. A secondary ossification center outlined by a cluster of red structures (blue circle; osteoid) was also detected at this time point. The corresponding zone in NCCSs, contained isolated cells resembling chondrocytes and chondroblasts embedded into a native pale-yellow matrix, with moderate acidification appearing as green around the lacunae (Fig. 3A-iii, inlet black arrowheads).
In the DZ, stratified columnar chondrocytes observed in NCCSs were replaced by highly mineralized (green) elongated hypertrophic cell columns in cultured femur slices (Fig. 3A-v, vi), and large multinucleated cells were detected at the metaphyseal junction (blue arrows). In the diaphysis, condensation and expansion of the bone collar were associated with a loss of marrow components (Fig. 3A-vii, black arrows) and detection of the mineralized new bone matrix (black arrows; pale yellow staining). NCCSs, however, exhibited a homogeneous distribution of bone marrow components (red clusters), as well as multinucleated osteoclast-like cells (Fig. 3A-vii, blue arrows).
Osteoclast-mediated bone remodeling in cultured femur slices was evaluated by tartrate-resistant acid phosphatase (TRAP) labeling. Although TRAP expression was barely detectable in cultured slices, a noticeable increase in mineralized bone matrix (black arrows), indicative of osteoclast-mediated resorption, was observed (Fig. 3B-ii). A similar pattern of TRAP expression and an increase in bone mineralized bone matrix were also detected in the diaphysis (Fig. 3B-iii, iv). In contrast, NCCSs exhibited TRAP-positive cells (blue arrows) in the diaphysis and metaphyseal junction with limited mineralized bone matrix (Fig. 3B-i).
Tissue mineralization during chondrogenic and osteogenic differentiation in cultured femur slices
Alizarin red staining confirmed a clear time-dependent increase in calcification in distinct zones of the epiphysis in cultured femur slices (Fig. 4A-i to iv). Interestingly, pronounced calcification was observed on day 15 in the regions of perichondrium (black arrows) at the metaphyseal junction (Fig. 4A-iv). Calcification of the bone collar was similarly also prominent in the diaphysis at this time point (Fig. 4A-v to viii). Quantifications performed on days 3 (20.39% ± 1.61%), 7 (26.85% ± 0.99%), and 15 (28.41% ± 1.02%) and in NCCSs (15.99% ± 0.74%) demonstrated the continuous increase of calcium deposits on days 7 and 15 with respect to NCCSs in both the bone collar and the metaphyseal junction, indicative of higher calcium turnover and tissue mineralization (Fig. 4B).

OTC cultures displayed increased calcium turnover and bone minerals.
Bone mineralization is defined by a unique combination of distinct bone-forming elements. The composition of these elements was assessed in cultured bone slices at different time points by ATR-IR spectral analysis (Fig. 4C). All essential peaks corresponding to hydroxyapatite/carbonates, collagen, and glycosaminoglycans (GAGs) were observed at the wave numbers of the corresponding elements (Table 1).
Crystallinity Index of Bone Characterized by ATR-IR Spectroscopy
GAGs, glycosaminoglycans.
Prominent peaks include, for example, v4, PO43− (553 cm−1), C-O stretching vibration of hyaluronic acid and chondroitin sulfate (1082 cm−1), amide III C-N stretching (1235 cm−1), amide II (1650 cm−1), as well as amide B (3283 cm−1). A time-dependent increase in all bone matrix elements, which peaked on day 15, was observed in the epiphysis, metaphysis, and diaphysis, indicating on-going mineralization. The peaks of the respective elements corresponded to the peaks determined for NCCSs and did not shift during culture. A slight variation in mineral composition was, however, observed in the epiphysis and metaphysis on day 15 (Fig. 4C, spectrum 1). The appearance of CO32− at 930 cm−1 indicates the formation of type-B apatite as compared with a potential type-A apatite in the diaphysis at this time point. The relevance of these observations is not entirely clear. However, type-B apatites are considered more labile and might influence solubility, shape, and size of apatite crystallites.
Chondrogenic and osteogenic protein expression in cultured femur slices
To verify chondrogenic and osteogenic differentiation in cultured femur slices detected by histological staining, expression of protein markers of chondrogenic differentiation (collagen II), chondrocyte maturation and early osteogenesis (Runx2), and late osteogenesis (osteocalcin) was evaluated in 15-day cultured slices by immunohistochemistry (Fig. 5A–E). Respective negative controls with primary antibody omission are presented in Supplementary Figure S4. Low level collagen II expression in NCCSs was detected in a diffused pattern across the epiphysis (Figs. 5A-i and 6-i). Higher collagen II expression was observed in cultured slices on day 15 surrounding the growth plate; however, the expression diminished at the central epiphysis, indicating an initiation of ossification in these areas (Figs. 5A-ii and 6-i). Pertaining to epiphysis, collagen II expression was notably reduced in the TZ (23.31% ± 3.81% vs. 10.66% ± 1.76%) compared with NCCSs (Figs. 5A-iii to iv and 6-i).
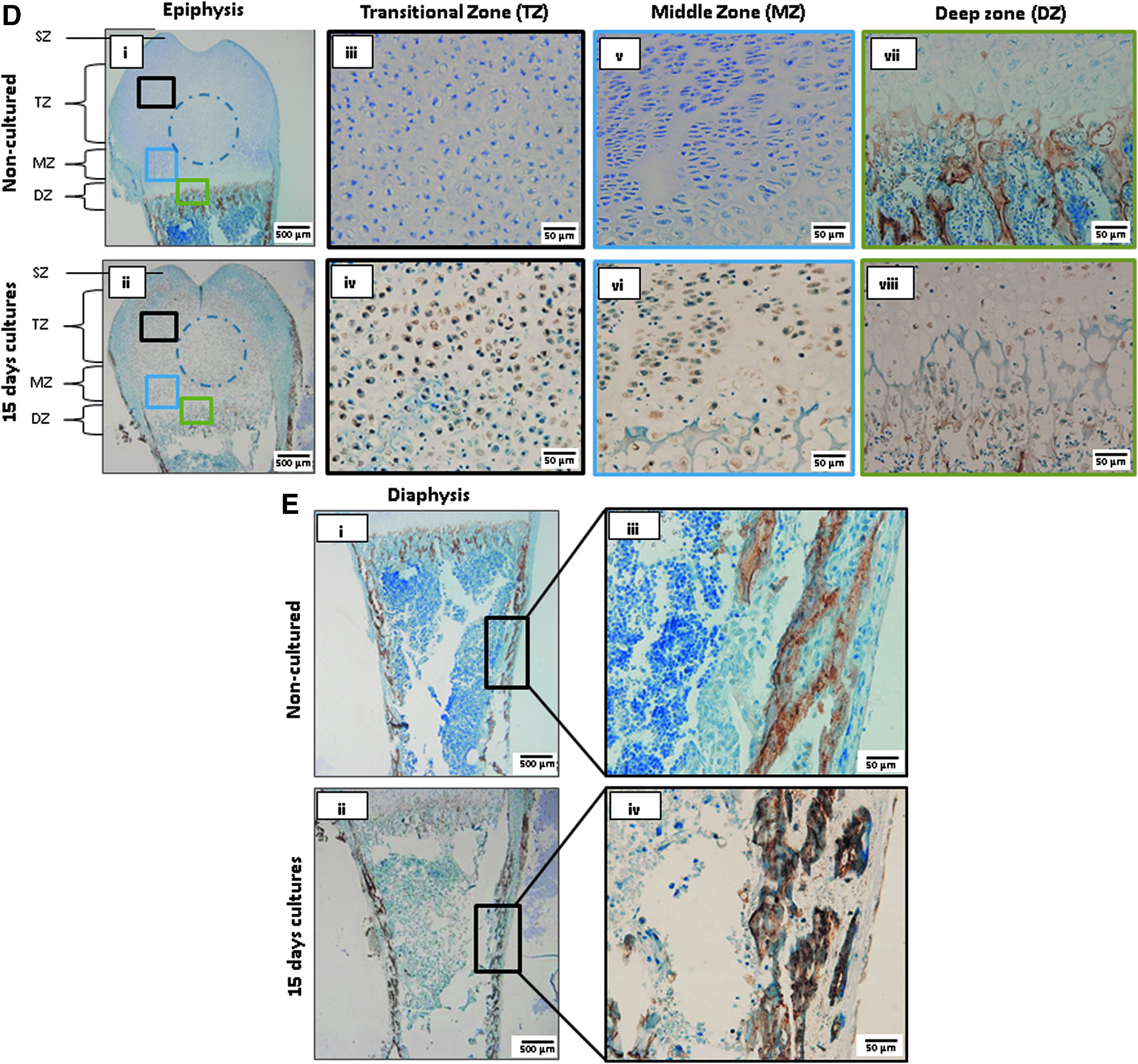
Expression of early and late chondrogenic and osteogenic differentiation markers indicated advanced endochondral transition.
In the MZ and DZ, collagen II expression was elevated compared with NCCSs (MZ: 19.06% ± 1.15% vs. 67.62% ± 1.19% and DZ: 21.95% ± 3.70% vs. 58.96% ± 3.89%; Figs. 5A-v to viii and 6-i). Contrary to collagen II expression, an increase in Runx2 expression was noticed around the center of the epiphysis at locations almost devoid of collagen II expression (Figs. 5B-i to ii and 6-ii). Our observations indicated overall higher Runx2 expression in the epiphysis corresponding to TZ, MZ, and DZ on day 15 compared with NCCSs (TZ: 5.46% ± 1.36% vs. 32.67% ± 1.71%; MZ: 8.21% ± 1.15% vs. 27.77% ± 2.16%; DZ: 14.23% ± 2.04% vs. 33.98% ± 1.93%; Figs. 5B-iii to viii and 6-ii). Interestingly, these changes were relatable to caspase 3 expression in 15-day slice cultures (Supplementary Fig. S5).

Quantification of immunostaining. Quantification of
Runx2 expression was also higher in the diaphysis and cortical bone than in NCCSs (16.98% ± 1.51% vs. 29.58% ± 2.85%; Figs. 5C-i to iv and 6-ii). This robust manifestation of osteogenic differentiation was further substantiated by osteocalcin staining, apeptide hormone secreted by osteoblasts, and incorporated within the calcified bone matrix. On day 15, a higher number of osteocalcin expressing cells were, accordingly, observed in all epiphyseal zones (Figs. 5D-i to viii and 6-iii) and in the diaphysis (Figs. 5E-i to iv and 6-iii) than in NCCSs (TZ: 1.03% ± 0.28% vs. 17.24% ± 1.75%; MZ: 1.08% ± 0.21% vs. 17.45% ± 1.67%; DZ: 12.60% ± 2.28% vs. 19.54% ± 1.64%; cortical bone: 45.66% ± 0.89% vs. 58.04% ± 3.13%).
Discussion
In this study we have demonstrated a methodology to obtain precision-cut 300 μm sagittal femur slices from 4-day-old rats comprising epiphysis, growth plate, and meta-diaphysis that can undergo endochondral and osteogenic bone development ex vivo. Our study was prompted by studies with other tissues and organs that demonstrate that slices of this thickness are sufficiently perfused by gases and nutrients during static culture to remain metabolically active for experimentally meaningful time periods.31,32 To validate our technique, we have cultured the resultant 300 μm femoral slices in an osteogenic medium and characterized endochondral osteogenic development. Our study demonstrated orchestrated cellular transitions of bone cells performing endochondral ossification, along with subsequent increase in chondrogenic and osteogenic markers, coupled with mineralization in both epiphysis and diaphysis of cultured slices compared with NCCSs.
Endochondral differentiation is manifested by expression and localization of chondrogenic marker type II collagen in the growth plate of the epiphysis33,34 with simultaneous increase in osteogenic markers 35 Runx2, a key transcription factor in osteogenesis,36–39 and bone matrix protein osteocalcin.40,41
In 15-day cultured slices, we observed a strong localization of type II collagen to the epiphyseal transitional and proliferative zones, distinguishing immature and mature cartilage compared with controls, indicating progressive endochondral osteogenesis, as well as potential transdifferentiation of chondrocytes into osteoblasts, as reported in previous studies.42,43 Interestingly the collagen II matrix modulations from our study coincided with postnatal in vivo bone development shown by Lui et al. and Uchimura et al..44,45 Similar pattern of increase in Runx2 and osteocalcin expression was also indicated in several in vivo studies, illustrating bone development; however, we would like to point out that currently existing ex vivo models do not reveal such close resemblance to in vivo cellular transitions as shown in this particular slice model.
We also validated the mineralization profile of our cultures through quantitative stainings and ATR-IR spectroscopy, and observed that altered cellular phenotypes consecutively increased calcium deposits and bone minerals on days 3, 7, and 15 in vitro. These findings were also in line with several previous studies indicating bone development,46–49 thus inferring a nondestructive method of screening in our model.
Most studies opt for coculturing strategies to stimulate interaction of osteoclasts with other bone cells in ex vivo settings, nonetheless, our model evades this long existing challenge and provides a dynamic platform for interventional studies ex vivo. We confer that none of the existing ex vivo bone culture models supports the multidimensional interaction of physiological zones to understand the cellular dynamics involved in bone growth and development with such great accessibility. For instance, a proof of concept study by Saunders et al. 50 evaluated the feasibility of culturing whole femurs from postnatal day rats for up to 8 days and showed an increase in femur length, proteoglycans, mineralization, and housing of osteocytes and osteoblasts. Notably, the osteogenic properties of our slice cultures were far better validated than in this particular study through differential and immunostainings to capture zone-specific changes.
Attempts to establish organotypic bone cultures began as early as 1929 to gain insights into longitudinal bone development, although most studies were confined to embryonic tissues potentially due to limited diffusion of extracellular molecules to thick calcified bone tissue.51,52 Previous studies have utilized rodent bone organotypic culture models from embryonic metatarsals, whole femurs, mandible slices, femoral heads, calvarias, and trabecular cores to investigate factors influencing bone growth and metabolism. 17 Notably, most studies used murine embryonic metatarsals and chick embryos to study endochondral transitions53,54; given the immaturity of these tissues, their acceptability is debatable due to developmental discrepancies.14,55 Farquharson and Jefferies 56 and Nowlan et al., 57 for instance, noted the absence of a secondary ossification center, lack of vascular supply, and growth plate variations as significant caveats of embryonic avian bones. 58
Although a number of studies conducted on murine metatarsals and embryonic chick femurs have significantly contributed in understanding the skeletal morphogenesis,59–61 they fail to capture the interaction of (cartilage) epiphysis and meta-diaphysis (bone marrow) due to sparsely developed physiological zones (epiphysis and diaphysis) that are imperative for longitudinal bone formation. A recent review by Kohli et al. discussed that only a few negligible 3D models were partly capable of demonstrating synchronized metamorphosis of osteocytes, osteoblasts, and osteoclasts that are vital for orchestrated skeletal development and maintenance of signaling gradients.62,63
We believe this can be better achieved in our model as it comprises slightly advanced zone-specific mesenchyme and has the natural tendency to activate resorption, whereas embryonic bones contain poorly defined zones that complicate in vivo correlation. In our model we have successfully recapitulated the functionality of both new bone formation and osteoclast-mediated resorption in both metaphysis and diaphysis in a manner similar to in vivo events as indicated by TRAP staining. 64 Although organ slice cultures facilitate the study of bone–cell interactions and their underlying mechanisms, due to a lack of vascularization, their growth and development are still not comparable with in vivo studies.35,65 Most studies adapt perfusion bioreactors to achieve adequate nutrient supply in bone explant cultures. 66
Our bone slice culture model, in contrast, provides a better interface for nutrient diffusion catering to all core bone components responsible for linear growth, and thereby allows, in its most basic format, for simultaneous capture of both specific effects on growth plate dynamics and new bone formation. Moreover, their enhanced interaction and accessibility make them a unique and powerful tool for adapting coculture strategies and investigating osteolytic bone cancers and bone metastasis.67,68
To our knowledge, a reliable basic bone model able to improve our understanding of cellular manifestations involved in the growth and development of longitudinal bone is currently unavailable. 69 Our bone slice culture could both fill this gap to a large extent and, moreover, be utilized as a platform to explore numerous possibilities to in the area of bone repair and regeneration.61,70–73 From an ethical standpoint, it could also impact the 3R principles of refining animal experiments by providing the basis for a reliable culture system that could perhaps relate to in vivo experiments.74,75
Despite its robustness, the absence of vascularization and systemic influences will need to be taken into account when interpreting results, although it has been suggested that these factors may not be crucial for short-term bone developmental studies. 76 The other known limitations of our model may incur difficulties in mechanical stimulation, limited cells, and limited lifespan, although the tailoring strategies are still to be explored. In addition, integrating some of the high-throughput nucleotide sequencing, translational profiling to assess linear growth, and epigenetic investigations could contribute significantly to future applications.77–79
Conclusion
To summarize, we have successfully demonstrated a methodology to generate 300 μm organotypic femur slice culture model capable of demonstrating endochondral morphological transitions similar to a developing bone. Their unique accessibility makes them a great platform to unravel some of the complex biological events underlying bone development and to enable the screening of concurrent effects of pharmacological agents, biomaterials, and regenerative cells on endochondral bone development. This basic methodology can further be extrapolated to derive better insights into the mechanisms underlying a wide range of metabolic bone syndromes and bone traumas.32,80,81
Footnotes
Acknowledgments
This research was supported by Marie Skłodowska-Curie Actions of the European Union's Seventh Framework Programme FP7/2007-2013/under REA grant agreement number 289163; by CAMed (COMET K-Project 871132), funded by Austrian Federal Ministry of Transport, Innovation and Technology (BMVIT) and the Austrian Federal Ministry for Digital and Economic Affairs (BMDW) and the Styrian Business Promotion Agency (SFG); and by the University Research Grant (Triennial Research Plan 2016–2018), Department of Biomedical and Biotechnological Sciences (BIOMETEC), University of Catania, Italy. This research work is a part of doctoral dissertation at Medical University of Graz, Austria. The authors extend special thanks to Dr. Muammer Üçal and Dr. Silke Patz for their technical support and guidance. Authors also thank Ing. Emilio Gomez for his help in animal care and handling and Mag. Klaus Kraitsy for his help to set up the vibratome device. The authors also thank Dr. Chirs Wrighton for extending his help in scientific editing and proof reading this article.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.